

Forschung

| Fakultät für Biologie und Vorklinische Medizin |
| Institut für Anatomie |
| Lehrstuhl für Molekulare und Zelluläre Anatomie |
| Forschung |
 |  |  |  |
Forschungsschwerpunkte
Im Zentrum der wissenschaftlichen Tätigkeit steht die Aufklärung der Mechanismen, die zum akuten und chronischen Nierenversagen führen. Als Modelle dienen dabei zwei Erbkrankheiten, nämlich das Nagel-Patella-Syndrom und die polyzystische Nierenerkrankung, die wie die erworbenen Formen zum einen die Podozyten und zum anderen die Nierentubuli betreffen. Die (langfristige) Hoffnung geht dahin, daß ein besseres Verständnis der Krankheitsentstehung zu einer rationelleren und damit wirksameren Entwicklung von Therapien führt.
Pathogenetische Vorgänge folgen oft einem komplexen Ablauf und sind wohl am besten über einen vielschichtigen Forschungsansatz aufzuklären. Das Institut für Molekulare und Zelluläre Anatomie bemüht sich deshalb, durch die konsequente Kombination molekularbiologischer, zellbiologischer, biochemischer und morpholo-gischer Techniken die entsprechenden Fragen anzugehen (siehe unten).
Nagel-Patella-Syndrom
Die Nieren als Ort der Harnproduktion
Zusammen mit der Leber sind die beiden Nieren die maßgeblichen Organe für die Ausscheidung von Stoffwechselprodukten und Giften. Trotz der Tatsache, dass eine Niere nur etwa faustgroß ist, erhalten die Nieren ~20% des vom Herzen ausgeworfenen Blutvolumens. Die Nieren stellen einen sehr wirksamen Filter dar, über den kleine Stoffe wie Stoffwechselprodukte, Salze oder Medikamente ausgeschieden, die meisten der wertvollen Eiweißmoleküle allerdings zurückgehalten werden. Für die Filterfunktion der Nieren sind insbesondere die "Füßchenzellen" in der Niere, die Podozyten, verantwortlich, welche bei verschiedensten Erbkrankheiten geschädigt sein können. Im Laufe eines Tages werden von den Nieren 180 Liter des sogenannten Primärfiltrats produziert, von dem bekanntermaßen allerdings nur ein oder zwei Liter ausgeschieden werden. Hierfür wiederum ist ein Röhrensystem in den Nieren verantwortlich, die sogenannten Nierentubuli.
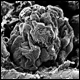
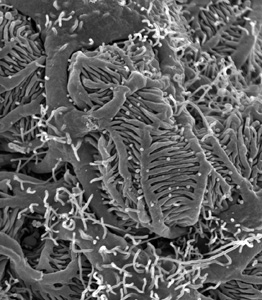 | Rasterelektronenmikroskopische Aufnahme der Füßchenzellen in der Niere (Podozyten). Deutlich sind die größeren Primärfortsätze sowie die vielen feinen Fußfortsätze zu erkennen. |
Klinische Aspekte
Die am Nagel-Patella-Syndrom leidenden Patienten sind relativ leicht anhand ihrer fehlgebildeten Fingernägel und mißgebildeten oder gar fehlenden Kniescheiben zu diagnostizieren, deshalb auch der Name. Es handelt sich um eine autosomal-dominant weitergegebene Erbkrankheit, die nur selten beobachtet wird. Bemerkenswerterweise wurde bei bis zu 40% aller Patienten eine sich langsam entwickelnde Fehlfunktion der Nieren beschrieben, die von einer leichten Eiweißausscheidung bis zum kompletten Funktionsverlust der Nieren führen kann (Sweeney et al., 2003). Ursächlich hierfür ist ein Podozytenschaden, der sich in einem Verlust der Fußfortsätze und in charakteristischen Veränderungen der glomerulären Basalmembran äußert. Des Weiteren scheint das Nagel-Patella-Syndrom mit einem höheren Risiko verbunden zu sein, am grünen Star (Glaukom) zu erkranken (Bongers et al., 2002; Bongers et al., 2005; Sweeney et al., 2003). Wahrscheinlich hängt dieser Befund damit zusammen, dass das entsprechende Gen LMX1B auch bei der Entstehung des vorderen Augenabschnitts beteiligt ist (Pressman et al., 2000).
Aufgrund der Seltenheit des Nagel-Patella-Syndroms – in der wissenschaftlichen Literatur ist regelmäßig eine Häufigkeit von 1:50.000 zu lesen, ohne dass entsprechende Untersuchungen hierzu vorliegen – existieren keine klinischen Studien zu möglichen therapeutischen Maßnahmen. Erstaunlicherweise scheint ein Leben ohne Kniescheiben mit keinen größeren Beeinträchtigungen verbunden zu sein, so dass als wesentliche Symptomatik die Fehlfunktion der Nieren und das Glaukom berücksichtigt werden muss. Da auch hier keine 100%ige Korrelation beobachtet wird (bis zu 40% der Patienten entwickeln eine milde bis schwere Fehlfunktion der Nieren, bis zu 20% leiden an einem hohen Augeninnendruck oder sogar einem Glaukom) sind wahrscheinlich erst einmal nur routinemäßige Kontrollen von Nöten. Erst bei tatsächlich auftretenden Beschwerden müssen entsprechende therapeutische Anstrengungen unternommen werden, die leider nicht kausal angreifen, sondern nur die Symptome behandeln.
Genetik und Funktion des Proteins
Im Jahr 1998 wurden Mutationen im LMX1B Gen als krankheitsverursachend identifiziert (Dreyer et al., 1998; McIntosh et al., 1998; Vollrath et al., 1998). Dieses Gen kodiert für einen Transkriptionsfaktor, der eine wichtige Rolle nicht nur bei der Gliedmaßenentwicklung spielt (deshalb die Nagel- und Kniescheibensymptomatik), sondern auch bei der Ausbildung der Nieren. In den Nieren sind speziell die Podozyten betroffen, die bei der Filtration des Blutplasmas so wichtigen “Füßchenzellen”, denn den Podozyten von Lmx1b Knockout-Mäusen fehlen die Fußfortsätze und die Schlitzmembranen (Miner et al., 2002; Rohr et al., 2002). Eine wichtige Erkenntnis, die zu einem genaueren Verständnis der Wirkungsweise von LMX1B führen sollte, liegt in der Tatsache begründet, daß LMX1B zwei Gene reguliert, die bei anderen erblichen Nierenerkrankungen mutiert sind. Es sind dies die für Kollagen IV (Morello et al., 2001) und Podocin (Miner et al., 2002; Rohr et al., 2002) kodierenden Gene, in denen Mutationen bei Patienten mit Alport-Syndrom bzw. steroidresistentem nephrotischen Syndrom beschrieben wurden.
Der endgültige Nachweis, dass der Transkriptionsfaktor LMX1B in den Nieren ausschließlich die Funktion der Podozyten beeinträchtigt, stammt von Untersuchungen bei Mäusen, in denen das Lmx1b Gen ausschließlich in den Podozyten inaktiviert wurde. Hierbei stellte sich heraus, dass Lmx1b sowohl bei der Entstehung von Podozyten im fetalen Organismus (Suleiman et al., 2007) als auch bei der Aufrechterhaltung der Podozytenfunktion im adulten Organismus (Burghardt et al., 2013) von essentieller Bedeutung ist. Die derzeitige Arbeitshypothese geht davon aus, dass das Aktinzytoskelett bei einer Fehlfunktion von LMX1B seine spezifische Aufgabe nicht länger erfüllen kann (Burghardt et al., 2013).
Autosomal-dominante polyzystische Nierenerkrankung
Formenkreis der zystischen Nierenerkrankungen
Als Zysten bezeichnet man mit Flüssigkeit gefüllte und von einem Epithel ausgekleidete Hohlräume. Sie kommen nicht nur in den Nieren, sondern auch zum Beispiel in der Leber oder in der Bauchspeicheldrüse vor. Einzelne Nierenzysten besitzen in aller Regel keinen Krankheitswert und werden gar nicht so selten als Zufallsbefund nachgewiesen. Bei einem gehäuften Auftreten von Zysten spricht man demgegenüber von Zystennieren, sie sind ein wichtiges, oft sogar die Prognose bestimmendes Symptom eines ganzen Blumenstraußes verschiedener genetischer Erkrankungen. Hierzu gehören unter anderem die Nephro-nophthise als häufigste Ursache des Nierenversagens bei Kindern, das Bardet-Biedl-Syndrom, die tuberöse Sklerose, das Joubert-Syndrom, das Meckel-Gruber-Syndrom und die autosomal-rezessive und autosomal-dominante polyzystische Nierenerkrankung. Je nachdem, welche anderen Symptome bei den Patienten auftreten (diese können sehr heterogen sein wie Fettleibigkeit, geistige Retardierung oder eingeschränkte Sehfähigkeit), werden die Patienten den entsprechenden Syndromen zugeordnet – eine wichtige Voraussetzung für eine mögliche genetische Diagnostik.
| Polyzystische Niere einer 48 Jahre alten Frau. Während eine normale Niere nur etwa 150 g wiegt, kann eine polyzystische Niere ein Gewicht von über 4 kg erreichen! (freundlichst überlassen von N. Gassler) | 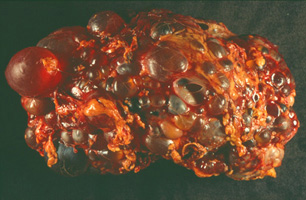 |
Klinische Aspekte
Die dominante Form ist mit einer Prävalenz von ~1:1.000 eine der häufigsten monogenetischen Erkrankungen des Menschen. Patienten mit autosomal-dominanter polyzystischer Nierenerkrankung erfahren oft erst im zweiten oder dritten Lebensjahrzehnt von ihrer Krankheit, zum Beispiel im Rahmen einer anderweitig durchgeführten Ultraschalluntersuchung. Ungefähr 50% der Patienten werden bis zum 60. Lebensjahr das Endstadium des chronischen Nierenversagens erreichen (Churchill et al., 1984; Gabow et al., 1992; Parfrey et al., 1990), sie repräsentieren damit knapp 10% aller dialysepflichtigen Patienten. Erstaunlicherweise ist es so, dass Patienten, die an einer Mutation im PKD1 Gen leiden, eine schlechtere Prognose besitzen als Patienten mit Mutationen im PKD2 Gen: Es kommt früher zum terminalen Nierenversagen, so dass die Patienten dialysepflichtig werden oder eine neue Niere benötigen.
Bedauerlicherweise existiert nach wie vor keine kausale Therapie bei den polyzystischen Nierenerkrankungen. Aufgrund ihrer Häufigkeit wurden bislang nur Studien für die autosomal-dominante Form durchgeführt. Während sich die Hoffnungen in Therapiestudien mit Everolimus (Walz et al., 2010) und Sirolimus (Serra et al., 2010), zweier auch als Immunsuppressiva eingesetzter Medikamente, zerschlugen, stimmen die Studienergebnisse mit Tolvaptan optimistischer (Torres et al., 2012). Bei Tolvaptan handelt es sich um ein Medikament, das die Wirkung des antidiuretischen Hormons (auch Vasopressin genannt) an seinem Rezeptor in der Niere blockiert. Dadurch soll eine Signalkaskade gehemmt werden, die auch das Wachstum von Nierenzysten befördert. Daraus erklärt sich eine der Nebenwirkungen von Tolvaptan, nämlich die erhöhte Harnproduktion, außerdem kann die Einnahme von Tolvaptan zur Beeinträchtigung der Leberfunktion führen.
Genetik und Funktion der Proteine
In den Jahren 1994 (European Polycystic Kidney Disease Consortium, 1994) und 1996 (Mochizuki et al., 1996) wurde von der Klonierung der bei diesen Patienten mutierten Gene berichtet, PKD1 und PKD2 genannt, womit nun prinzipiell eine Diagnosestellung auf molekularer Ebene möglich ist.
Das PKD1 Gen ist bei etwa 85% aller Patienten mit autosomal-dominanter polyzystischer Nierenerkrankung mutiert. Es kodiert für Polycystin-1, ein sehr großes Protein mit einer Länge von 4.302 Aminosäuren (Hughes et al., 1995; International Polycystic Kidney Disease Consortium, 1995). Aufgrund seiner Größe und des Vorhandenseins eng verwandter Gene kann sich das Auffinden der Mutation bei PKD1 Patienten schwierig gestalten. Die Funktion von Polycystin-1 ist bisher nur sehr unvollständig verstanden, weshalb nur darüber spekuliert werden kann, wie Mutationen zur Entstehung von Zysten führen. Möglicherweise ist Polycystin-1 bei der Ausbildung von Zell-Zell- und/oder Zell-Matrix-Kontakten beteiligt, hierfür gibt es bisher aber nur vereinzelte Hinweise. Allerdings wird gemeinhin akzeptiert, daß Polycystin-1 mit Polycystin-2 interagieren kann, dem von PKD2 kodierten Protein (Newby et al., 2002; Qian et al., 1997; Tsiokas et al., 1997).
Mutationen im PKD2 Gen wurden bei der Mehrzahl der restlichen Patienten, die an der autosomal-dominanten Form leiden, beschrieben. Das Polycystin-2 Protein des Menschen ist mit einer Länge von 968 Aminosäuren wesentlich kleiner als Polycystin-1. Deshalb und aufgrund des Fehlens sehr ähnlicher Gene ist PKD2 einer genetischen Diagnose wesentlich einfacher zugänglich als PKD1. Wahrscheinlich ist Polycystin-2 ein neuartiger Kationenkanal mit einer sehr hohen Leitfähigkeit von über 100 pS (González-Perrett et al., 2001; Hanaoka et al., 2000; Koulen et al., 2002; Vassilev et al., 2001), wobei aber kontrovers diskutiert wird, ob das Protein in der Zellmembran (Hanaoka et al., 2000; Luo et al., 2003) oder im endoplasmatischen Retikulum lokalisiert ist (Cai et al., 1999; Koulen et al., 2002).
Die Bildung von Zysten – primäre Zilien im Mittelpunkt der Wissenschaft
Wie entstehen nun Zysten? Genetisch spricht sehr viel dafür, daß jeweils beide Allele von PKD1 und PKD2 inaktiviert sein müssen, damit sich Zysten bilden (Pei, 2001). In Analogie zu den Tumorsuppressorgenen geht man zur Zeit davon aus, daß das in der Keimbahn gesunde Allel im Laufe des Lebens eine Mutation erleidet, die betroffenen Zellen erhalten möglicherweise einen Wachstumsvorteil und die Zystenbildung beginnt, weil ihr “Stopsignal” ausgefallen ist. In diesem Zusammenhang sei erwähnt, daß die Nierentubuli aus einem bindegewebigen Vorläufergewebe differenzieren, das heißt das Lumen der Tubuli entwickelt sich erst im Laufe der Zeit. Zu einem genau definierten Zeitpunkt muß die Lumenbildung allerdings zu einem Ende kommen, denn die Nierentubuli sind vergleichbar weit! Wenn das Stopsignal nicht mehr existiert, wird mehr Lumen gebildet und es entstehen Zysten (Gallagher et al., 2000).
Über die Natur des Stopsignals oder Sensors für die Weite der Nierentubuli gibt es nur vage Vorstellungen, möglicherweise kommt primären Zilien, fingerförmigen Ausstülpungen der Zellmembran, eine maßgebliche chemo- und/oder mechanosensorische Rolle zu. Die dafür sprechenden Befunde sind der Nachweis verschiedenster Zystenproteine in primären Zilien (Yoder et al., 2002) und der Verlust ihrer mechanosensorischen Eigenschaften bei Ausfall von Polycystin-1 und -2 (Nauli et al., 2003). Andere Untersuchungen wiederum weisen primären Zilien chemosensorische Eigenschaften zu, insbesondere sind hier Experimente bei Modellorganismen zu nennen. Wie oben bereits erwähnt, entsteht der Tubulusapparat der Niere aus einer bindegewebigen Vorläuferstruktur, mit anderen Worten müssen Hohlräume einer bestimmten Weite angelegt werden. Wie der dabei beteiligte Regelkreis ausschaut, ist vollkommen unverstanden. Vorstellbar ist zum Beispiel, dass die Weite des Lumens über die Flussgeschwindigkeit der Flüssigkeit in den Nierentubuli gemessen wird: Bei einer hohen Flussgeschwindigkeit (in engen Tubuli) wird das primäre Zilium stärker abgeknickt, bei einer niedrigen Flussgeschwindigkeit (in weiten Tubuli) dagegen weniger. Gegen dieses auf den ersten Blick sehr plausible Modell können verschiedene theoretische Einwände erhoben werden, so dass bislang nicht entschieden ist, ob eher die mechano- oder die chemosensorische Funktion von primären Zilien ausschlaggebend ist (Witzgall, 2005).
Literatur
Bongers, E.M.H.F., Gubler, M.-C., and Knoers, N.V.A.M. (2002). Nail-patella syndrome. Overview on clinical and molecular findings. Pediatr Nephrol 17, 703-712.
Bongers, E.M.H.F., Huysmans, F.T., Levtchenko, E., de Rooy, J.W., Blickman, J.G., Admiraal, R.J.C., Huygen, P.L.M., Cruysberg, J.R.M., Toolens, P.A.M.P., Prins, J.B., et al. (2005). Genotype-phenotype studies in nail-patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur J Hum Genet 13, 935-946.
Burghardt, T., Kastner, J., Suleiman, H., Rivera-Milla, E., Stepanova, N., Lottaz, C., Kubitza, M., Böger, C.A., Schmidt, S., Gorski, M., et al. (2013). LMX1B is essential for the maintenance of differentiated podocytes in adult kidneys. J Am Soc Nephrol 24, 1830-1848.
Cai, Y., Maeda, Y., Cedzich, A., Torres, V.E., Wu, G., Hayashi, T., Mochizuki, T., Park, J.H., Witzgall, R., and Somlo, S. (1999). Identification and characterization of polycystin-2, the PKD2 gene product. J Biol Chem 274, 28557-28565.
Churchill, D.N., Bear, J.C., Morgan, J., Payne, R.H., McManamon, P.J., and Gault, M.H. (1984). Prognosis of adult onset polycystic kidney disease re-evaluated. Kidney Int 26, 190-193.
Dreyer, S.D., Zhou, G., Baldini, A., Winterpacht, A., Zabel, B., Cole, W., Johnson, R.L., and Lee, B. (1998). Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet 19, 47-50.
European Polycystic Kidney Disease Consortium (1994). The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 77, 881-894.
Gabow, P.A., Johnson, A.M., Kaehny, W.D., Kimberling, W.J., Lezotte, D.C., Duley, I.T., and Jones, R.H. (1992). Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 41, 1311-1319.
Gallagher, A.R., Obermüller, N., Cedzich, A., Gretz, N., and Witzgall, R. (2000). An ever-expanding story of cyst formation. Cell Tissue Res 300, 361-371.
González-Perrett, S., Kim, K., Ibarra, C., Damiano, A.E., Zotta, E., Batelli, M., Harris, P.C., Reisin, I.L., Arnaout, M.A., and Cantiello, H.F. (2001). Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc Natl Acad Sci USA 98, 1182-1187.
Hanaoka, K., Qian, F., Boletta, A., Bhunia, A.K., Piontek, K., Tsiokas, L., Sukhatme, V.P., Guggino, W.B., and Germino, G.G. (2000). Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408, 990-994.
Hughes, J., Ward, C.J., Peral, B., Aspinwall, R., Clark, K., San Millán, J.L., Gamble, V., and Harris, P.C. (1995). The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 10, 151-159.
International Polycystic Kidney Disease Consortium (1995). Polycystic kidney disease: The complete structure of the PKD1 gene and its protein. Cell 81, 289-298.
Koulen, P., Cai, Y., Geng, L., Maeda, Y., Nishimura, S., Witzgall, R., Ehrlich, B.E., and Somlo, S. (2002). Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol 4, 191-197.
Luo, Y., Vassilev, P.M., Li, X., Kawanabe, Y., and Zhou, J. (2003). Native polycystin 2 functions as a plasma membrane Ca2+-permeable cation channel in renal epithelia. Mol Cell Biol 23, 2600-2607.
McIntosh, I., Dreyer, S.D., Clough, M.V., Dunston, J.A., Eyaid, W., Roig, C.M., Montgomery, T., Ala-Mello, S., Kaitila, I., Winterpacht, A., et al. (1998). Mutation analysis of LMX1B gene in nail-patella syndrome patients. Am J Hum Genet 63, 1651-1658.
Miner, J.H., Morello, R., Andrews, K.L., Li, C., Antignac, C., Shaw, A.S., and Lee, B. (2002). Transcriptional induction of slit diaphragm genes by Lmx1b is required in podocyte differentiation. J Clin Invest 109, 1065-1072.
Mochizuki, T., Wu, G., Hayashi, T., Xenophontos, S.L., Veldhuisen, B., Saris, J.J., Reynolds, D.M., Cai, Y., Gabow, P.A., Pierides, A., et al. (1996). PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272, 1339-1342.
Morello, R., Zhou, G., Dreyer, S.D., Harvey, S.J., Ninomiya, Y., Thorner, P.S., Miner, J.H., Cole, W., Winterpacht, A., Zabel, B., et al. (2001). Regulation of glomerular basement membrane collagen expression by LMX1B contributes to renal disease in nail patella syndrome. Nat Genet 27, 205-208.
Nauli, S.M., Alenghat, F.J., Luo, Y., Williams, E., Vassilev, P., Li, X., Elia, A.E.H., Lu, W., Brown, E.M., Quinn, S.J., et al. (2003). Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33, 129-137.
Newby, L.J., Streets, A.J., Zhao, Y., Harris, P.C., Ward, C.J., and Ong, A.C.M. (2002). Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J Biol Chem 277, 20763-20773.
Parfrey, P.S., Bear, J.C., Morgan, J., Cramer, B.C., McManamon, P.J., Gault, M.H., Churchill, D.N., Singh, M., Hewitt, R., Somlo, S., et al. (1990). The diagnosis and prognosis of autosomal dominant polycystic kidney disease. N Engl J Med 323, 1085-1090.
Pei, Y. (2001). A 'two-hit' model of cystogenesis in autosomal dominant polycystic kidney disease. Trends Mol Med 7, 151-156.
Pressman, C.L., Chen, H., and Johnson, R.L. (2000). Lmx1b, a LIM homeodomain class transcription factor, is necessary for normal development of multiple tissues in the anterior segment of the murine eye. Genesis 26, 15-25.
Qian, F., Germino, F.J., Cai, Y., Zhang, X., Somlo, S., and Germino, G.G. (1997). PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet 16, 179-183.
Rohr, C., Prestel, J., Heidet, L., Hosser, H., Kriz, W., Johnson, R.L., Antignac, C., and Witzgall, R. (2002). The LIM-homeodomain transcription factor Lmx1b plays a crucial role in podocytes. J Clin Invest 109, 1073-1082.
Serra, A.L., Poster, D., Kistler, A.D., Krauer, F., Raina, S., Young, J., Rentsch, K.M., Spanaus, K.S., Senn, O., Kristanto, P., et al. (2010). Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med 363, 820-829.
Suleiman, H., Heudobler, D., Raschta, A.-S., Zhao, Y., Zhao, Q., Hertting, I., Vitzthum, H., Moeller, M.J., Holzman, L.B., Rachel, R., et al. (2007). The podocyte-specific inactivation of Lmx1b, Ldb1 and E2a yields new insight into a transcriptional network in podocytes. Dev Biol 304, 701-712.
Sweeney, E., Fryer, A., Mountford, R., Green, A., and McIntosh, I. (2003). Nail patella syndrome: A review of the phenotype aided by developmental biology. J Med Genet 40, 153-162.
Torres, V.E., Chapman, A.B., Devuyst, O., Gansevoort, R.T., Grantham, J.J., Higashihara, E., Perrone, R.D., Krasa, H.B., Ouyang, J., Czerwiec, F.S., et al. (2012). Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367, 2407-2418.
Tsiokas, L., Kim, E., Arnould, T., Sukhatme, V.P., and Walz, G. (1997). Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc Natl Acad Sci USA 94, 6965-6970.
Vassilev, P.M., Guo, L., Chen, X.-Z., Segal, Y., Peng, J.-B., Basora, N., Babakhanlou, H., Cruger, G., Kanazirska, M., Ye, C.-p., et al. (2001). Polycystin-2 is a novel cation channel implicated in defective intracellular Ca2+ homeostasis in polycystic kidney disease. Biochem Biophys Res Commun 282, 341-350.
Vollrath, D., Jaramillo-Babb, V.L., Clough, M.V., McIntosh, I., Scott, K.M., Lichter, P.R., and Richards, J.E. (1998). Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum Mol Genet 7, 1091-1098.
Walz, G., Budde, K., Mannaa, M., Nürnberger, U., Wanner, C., Sommerer, C., Kunzendorf, U., Banas, B., Hörl, W.H., Obermüller, N., et al. (2010). Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 363, 830-840.
Witzgall, R. (2005). New developments in the field of cystic kidney diseases. Curr Mol Med 5, 455-465.
Yoder, B.K., Hou, X., and Guay-Woodford, L.M. (2002). The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 13, 2508-2516.
Angewendete Methoden (unvollständig)
Bindungsstellenselektion
“Branching morphogenesis” in dreidimensionalen Kollagengelen
CAT-Assay
Elektronenmikroskopie
Fluorographie 35S-markierter Proteine
Gel shift-Assay
Herstellung monoklonaler Antikörper
Herstellung polyklonaler Antikörper
Herstellung von Kernextrakten
Hybridisierung
Immunpräzipitation
Immunzytochemie
In situ-Hybridisierung
In vitro-Transkription/Translation
LDH-Assay
Luziferase-Assay
Metabolische Markierung
Mutagenese-Studien
Northern Blot
Organkulturen
PCR
Proteinaufreinigung über GST- und Ni2+-Säulen
Präparation von Plasmiden aus Bakterien im kleinen und großen Maßstab
Präparation von RNA
Random priming
RNase protection-Assay
Screening von Bibliotheken
Sequenzierung
Southern Blot
Southwestern Blot
Transfektion von Säugerzellen (transient und stabil)
Transformation von Bakterien und Hefe
Transgene Tiere
Western Blot
Zellproliferationsassays
Zweihybrid-Technologie
-->
Anita Hecht - 26.10.2023 12:56 