Research
Biochemistry II
Prof. Dr. Reinhard Sterner
|




Enzyme engineering
Tailoring enzyme activity, stability and quaternary structure
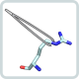
Generation of enzymes with a desired combination of structure, activity, and stability is the central goal of protein engineering. To achieve this goal we are using both directed evolution (the combination of random mutagenesis and selection or screening) and rational design (the introduction of specific amino acid exchanges on the basis of structural or mechanistic considerations). Directed evolution, for example, allowed us to significantly increase the low catalytic activity of a dimeric metabolic enzyme whereas rational design was used to generate a fully active monomeric variant of this enzyme. Based on these achievements, we are now performing sophisticated computer-based rational design to change the interaction specificities between the protein subunits from different enzyme complexes.
Light-control of enzymes
The targeted control of enzymes by light is a rapidly emerging field of protein design and synthetic biology. One such approach, which is pursued in cooperation with the group of Prof. Burkhard König (Organic Chemistry, University of Regensburg) is based on the use of photoswitchable small molecules that act as competitive enzyme inhibitors. The conformation of these photoswitches is modified by irradiation, which leads to a different affinities for the active site and thus allows us to fine-tune catalytic activity by light. Currently, we are using photo-responsive unnatural amino acids to light-control the propagation of an allosteric signal within a hetero-dimeric enzyme complex.
Enzyme evolution
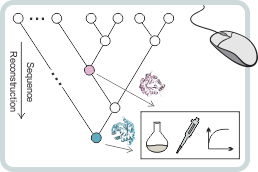
Ancestral sequence reconstruction
We are performing molecular paleontology to computationally calculate the amino acid sequences of proteins from extinct species. Following such ancestral sequence reconstruction (ASR), the corresponding primordial enzymes are produced and experimentally characterized. In cooperation with the group of Prof. Rainer Merkl (Biochemistry II, University of Regensburg), we have applied ASR to characterize an individual enzyme and an enzyme complex from the last universal common ancestor of all organisms (LUCA) or the last universal common ancestor of all bacteria (LBCA). These ancestral proteins were highly thermostable and catalytically active indicating that highly sophisticated enzymes and enzyme complexes existed already 3.5 billion years ago. We are now using ASR to identify residues that are crucial for the functional properties of various enzymes.
Evolutionary relationship between primary and secondary metabolism
It is generally assumed that enzymes from secondary metabolism have evolved from enzymes of primary metabolism. We have tested this hypothesis with the chorismate-utilizing enzymes anthranilate synthase (AS) from primary and isochorismate synthase (ICS) from secondary metabolism. Both enzymes catalyze mechanistically related reactions using ammonia and water as nucleophiles, respectively. We have found that the nucleophile specificity of AS can be extended from ammonia to water by just two amino acid exchanges in a channel leading to the active site. The observed ICS/AS bi-functionality demonstrates that a secondary metabolic enzyme can readily evolve from a primary metabolic enzyme without requiring an initial gene duplication event. We are now using similar approaches to interconvert other enzymes from primary metabolism to related ones from secondary metabolism.
Multi-enzyme complexes
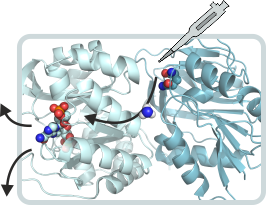
Glutamine amidotransferases
Glutamine amidotransferases are a family of enzymes whose members contain two active sites that are often located on two different polypeptide chains. The glutaminase subunit hydrolyses glutamine to glutamate and ammonia, which is added to a specific acceptor substrate at the active site of the synthase subunit. There are two important questions regarding the mechanism of glutamine amidotransferases: How are the activities at the two active sites allosterically coordinated? How is nascent ammonia transferred from the glutaminase to the synthase subunit without getting in contact with bulk water? We are investigating these questions on the examples of imidazole glycerol phosphate synthase (ImGPS) and aminodeoxychorismate synthase (ADCS). ImGPS and ADCS are key metabolic enzymes that link histidine and purine biosynthesis and catalyze the first step in folate biosynthesis, respectively.
Tryptophan synthase
The classical tryptophan synthase (TrpS) as found in most bacteria and plants is composed of α- und β-subunits. These subunits form a permanent αββα complex in which the reaction intermediate indole is channeled from the α- to the β-subunit. We have analyzed a novel type of TrpS complex, which is mainly found in Archaea. Interestingly, the relative orientation of the α- and β-subunits of the archaeal TrpS, its indole channel, and its allosteric activation mechanism differ from the “classical” bacterial and plant TrpS. Moreover, the substrate of the β-subunit is phosphoserine instead of serine. Thus, bacterial/plant and archaeal TrpS present an example for the convergent evolution of two isofunctional enzyme complexes.
A. Kneuttinger - 10.11.2020 09:43 
