

Research

Research interests |
| The research interests are focussed on the mechanisms leading to acute and chronic renal failure. We are using two hereditary diseases, polycystic kidney disease and nail-patella syndrome, as model systems, which as the acquired forms affect the renal tubuli and the podocytes, respectively. A better understanding of the phato- genetic events should ultimately result in more rational and efficient therapies.
More often than not those pathogenetic mechanisms are rather complex and only amenable to a multi-pronged approach. The Institute for Molecular and Cellular Anatomy therefore tries to combine molecular biology, cell biology, biochemical and morphological techniques (see below) in order to answer specific questions pertaining to this topic |
| Polycystic kidney disease | |
| Polycystic kidney disease can be inherited in an autosomal-dominant and autosomal-recessive fashion, furthermore acquired forms have been described which, however, will not be discussed further. At a prevalence of approximately 1 in 1,000 the dominant form presents one of the most frequent monogenetic diseases in man, while the recessive form is much less common (precise statistics do not exist); meanwhile the mutated genes have been identified for both forms. | |
| Polycystic kidney of a 48 year old woman. While a normal kidney only weighs about 150 g, a polycystic kidney can reach a weight of over 4 kg! (courtesy of N. Gassler). | 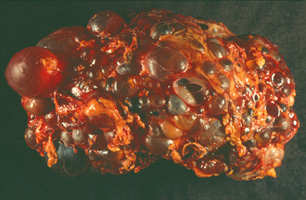 |
| It is not unusual that patients with autosomal-dominant polycystic kidney disease find out about their diagnosis only at age 20 or 30, for example when they have to undergo an ultrasound exam. Approximately 50% of the patients will reach end-stage renal failure until age 60, they therefore account for close to 10% of all patients on dialysis. In 1994 (European Polycystic Kidney Disease Consortium, 1994) and 1996 (Mochizuki et al., 1996) the positional cloning of the genes mutated in those patients (PKD1 and PKD2) was reported. Therefore at least in principle a diagnosis is now possible also on a molecular level. The PKD1 gene is mutated in approximately 85% of all patients with autosomal-dominant polycystic kidney disease. It encodes polycystin-1, a very large protein of 4.302 amino acids in length (Hughes et al., 1995; International Polycystic Kidney Disease Consortium, 1995). Because of its size and the presence of closely related genes finding mutations in PKD1 patients can turn out to be a difficult undertaking. The function of polycystin-1 so far is only incompletely understood, therefore it has to remain a matter of speculation how mutations lead to the development of cysts. Possibly polycystin-1 is a component of cell-cell and/or cell-matrix contacts, although there is only little supporting evidence for this hypothesis so far. It is, however, generally accepted that polycystin-1 interacts with polycystin-2, the protein encoded by the PKD2 gene (Newby et al., 2002; Qian et al., 1997; Tsiokas et al., 1997). Mutations in the PKD2 gene have been described in the majority of the remaining patients suffering from the autosomal-dominant form. At a length of 968 amino acids the polycystin-2 protein is much smaller than polycystin-1. For that reason and because closely related genes have not been described in the human genome mutations can be more easily detected in PKD2 than in PKD1. It is very likely that polycystin-2 represents a new class of cation channels with a large conductivity of >100 pS (González-Perrett et al., 2001; Hanaoka et al., 2000; Koulen et al., 2002; Vassilev et al., 2001), although there exists some controversy whether the protein is located in the plasma membrane (Hanaoka et al., 2000; Luo et al., 2003) or in the endoplasmic reticulum (Cai et al., 1999; Koulen et al., 2002). Autosomal-recessive polycystic kidney disease mostly affects patients in early childhood and carries with it a rather poor prognosis. In 2002 the affected gene was almost simultaneously identified by two groups using positional cloning (Onuchic et al., 2002; Ward et al., 2002). The PKHD1 gene also encodes a very large protein, which has been named fibrocystin and polyductin, respectively, and which probably exists in many different forms due to alternative splicing of the mRNA. The function of fibrocystin/polyductin is completely unknown, not even its intracellular location has been determined. How do cysts develop? There are a lot of genetic arguments that both alleles of PKD1 and PKD2 have to be inactivated before cysts can form (Pei, 2001). In analogy to tumor suppressor genes it is believed that the allele, which is not mutated in the germline, suffers a mutation at some point in life. The affected cells may gain a growth advantage and cyst formation starts because the “stop signal” is missing. In this context it has to be mentioned that the renal tubules develop from a mesenchymal precursor tissue, i.e. the tubular lumen forms over time. At a so for poorly defined timepoint the lumen formation has to stop, because all the tubules are equally wide. If the stop signal does not funtion any longer, then more lumen will form and cysts develop (Gallagher et al., 2000). The nature of the sensor is unknown, possibly primary cilia, finger-like protrusions of the plasma membrane, play a decisive chemo- and/or mechanosensory role. This notion is supported by the detection of various cyst-associated proteins in primary cilia (Yoder et al., 2002) and the loss of their mechanosensory capabilities upon the inhibition of polycystin-1 und -2 (Nauli et al., 2003). Cai, Y., Maeda, Y., Cedzich, A., Torres, V. E., Wu, G., Hayashi, T., Mochizuki, T., Park, J. H., Witzgall, R., and Somlo, S. (1999). Identification and characterization of polycystin-2, the PKD2 gene product. J. Biol. Chem. 274, 28557-28565. European Polycystic Kidney Disease Consortium (1994). The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 77, 881-894. Gallagher, A. R., Obermüller, N., Cedzich, A., Gretz, N., and Witzgall, R. (2000). An ever-expanding story of cyst formation. Cell Tissue Res. 300, 361-371. González-Perrett, S., Kim, K., Ibarra, C., Damiano, A. E., Zotta, E., Batelli, M., Harris, P. C., Reisin, I. L., Arnaout, M. A., and Cantiello, H. F. (2001). Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc. Natl. Acad. Sci. USA 98, 1182-1187. Hanaoka, K., Qian, F., Boletta, A., Bhunia, A. K., Piontek, K., Tsiokas, L., Sukhatme, V. P., Guggino, W. B., and Germino, G. G. (2000). Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408, 990-994. Hughes, J., Ward, C. J., Peral, B., Aspinwall, R., Clark, K., San Millán, J. L., Gamble, V., and Harris, P. C. (1995). The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 10, 151-159. International Polycystic Kidney Disease Consortium (1995). Polycystic kidney disease: The complete structure of the PKD1 gene and its protein. Cell 81, 289-298. Koulen, P., Cai, Y., Geng, L., Maeda, Y., Nishimura, S., Witzgall, R., Ehrlich, B. E., and Somlo, S. (2002). Polycystin-2 is an intracellular calcium release channel. Nat. Cell Biol. 4, 191-197. Luo, Y., Vassilev, P. M., Li, X., Kawanabe, Y., and Zhou, J. (2003). Native polycystin 2 functions as a plasma membrane Ca2+-permeable cation channel in renal epithelia. Mol. Cell. Biol. 23, 2600-2607. Mochizuki, T., Wu, G., Hayashi, T., Xenophontos, S. L., Veldhuisen, B., Saris, J. J., Reynolds, D. M., Cai, Y., Gabow, P. A., Pierides, A., Kimberling, W. J., Breuning, M. H., Deltas, C. C., Peters, D. J. M., and Somlo, S. (1996). PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272, 1339-1342. Nauli, S. M., Alenghat, F. J., Luo, Y., Williams, E., Vassilev, P., Li, X., Elia, A. E. H., Lu, W., Brown, E. M., Quinn, S. J., Ingber, D. E., and Zhou, J. (2003). Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33, 129-137. Newby, L. J., Streets, A. J., Zhao, Y., Harris, P. C., Ward, C. J., and Ong, A. C. M. (2002). Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J. Biol. Chem. 277, 20763-20773. Onuchic, L. F., Furu, L., Nagasawa, Y., Hou, X., Eggermann, T., Ren, Z., Bergmann, C., Senderek, J., Esquivel, E., Zeltner, R., Rudnik-Schöneborn, S., Mrug, M., Sweeney, W., Avner, E. D., Zerres, K., Guay-Woodford, L. M., Somlo, S., and Germino, G. G. (2002). PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 70, 1305-1317. Pei, Y. (2001). A 'two-hit' model of cystogenesis in autosomal dominant polycystic kidney disease. Trends Mol. Med. 7, 151-156. Qian, F., Germino, F. J., Cai, Y., Zhang, X., Somlo, S., and Germino, G. G. (1997). PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat. Genet. 16, 179-183. Tsiokas, L., Kim, E., Arnould, T., Sukhatme, V. P., and Walz, G. (1997). Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc. Natl. Acad. Sci. USA 94, 6965-6970. Vassilev, P. M., Guo, L., Chen, X.-Z., Segal, Y., Peng, J.-B., Basora, N., Babakhanlou, H., Cruger, G., Kanazirska, M., Ye, C.-p., Brown, E. M., Hediger, M. A., and Zhou, J. (2001). Polycystin-2 is a novel cation channel implicated in defective intracellular Ca2+ homeostasis in polycystic kidney disease. Biochem. Biophys. Res. Commun. 282, 341-350. Ward, C. J., Hogan, M. C., Rossetti, S., Walker, D., Sneddon, T., Wang, X., Kubly, V., Cunningham, J. M., Bacallao, R., Ishibashi, M., Milliner, D. S., Torres, V. E., and Harris, P. C. (2002). The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 30, 259-269. Yoder, B. K., Hou, X., and Guay-Woodford, L. M. (2002). The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 13, 2508-2516. | |
| Nail-patella-syndrome | |
| Patients suffering from nail-patella-syndrome can be rather easily diagnosed because of their dysplastic fingernails and their deformed or even missing patella, hence the name. It is a rare hereditary disease, which is inherited in an autosomaldominant fashion. Remarkably about a third to 50% of those patients slowly develop chronic renal failure. | |
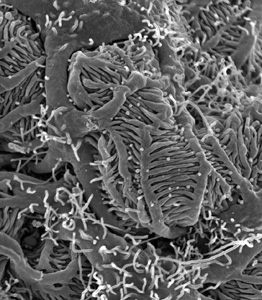 | Scanning electron micrograph of podocytes, a very special cell type only found in the kidney. One can easily see the large primary processes and the many fine foot processes. |
| In 1998 mutations in the LMX1B gene were identified in patients with nail-patella-syndrome (Dreyer et al., 1998; McIntosh et al., 1998; Vollrath et al., 1998). This gene codes for a transcription factor, which not only plays an important role in limb development, which is why the nails and the patella are affected, but also in the differentiation of the kidney. In the kidney it is the podocyte, the cell type crucially important for the filtration of the blood, which is affected by the mutations - podocytes of Lmx1b knockout mice are lacking foot processes and slit diaphragms (Miner et al., 2002; Rohr et al., 2002). An important finding, which should lead to a better understanding of LMX1B, stems from the fact that LMX1B regulates two genes, which are mutated in two other hereditary kidney diseases. These genes code for collagen IV (Morello et al., 2001) and podocin (Miner et al., 2002; Rohr et al., 2002) and are mutated in patients with Alport syndrome and steroid- resistant nephrotic syndrome, respectively. The mechanism, by which LMX1B regulates the differentiation of podocytes, is under intense investigation. Dreyer, S. D., Zhou, G., Baldini, A., Winterpacht, A., Zabel, B., Cole, W., Johnson, R. L., and Lee, B. (1998). Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat. Genet. 19, 47-50. McIntosh, I., Dreyer, S. D., Clough, M. V., Dunston, J. A., Eyaid, W., Roig, C. M., Montgomery, T., Ala-Mello, S., Kaitila, I., Winterpacht, A., Zabel, B., Frydman, M., Cole, W. G., Francomano, C. A., and Lee, B. (1998). Mutation analysis of LMX1B gene in nail-patella syndrome patients. Am. J. Hum. Genet. 63, 1651-1658. Miner, J. H., Morello, R., Andrews, K. L., Li, C., Antignac, C., Shaw, A. S., and Lee, B. (2002). Transcriptional induction of slit diaphragm genes by Lmx1b is required in podocyte differentiation. J. Clin. Invest. 109, 1065-1072. Morello, R., Zhou, G., Dreyer, S. D., Harvey, S. J., Ninomiya, Y., Thorner, P. S., Miner, J. H., Cole, W., Winterpacht, A., Zabel, B., Oberg, K. C., and Lee, B. (2001). Regulation of glomerular basement membrane collagen expression by LMX1B contributes to renal disease in nail patella syndrome. Nat. Genet. 27, 205-208. Rohr, C., Prestel, J., Heidet, L., Hosser, H., Kriz, W., Johnson, R. L., Antignac, C., and Witzgall, R. (2002). The LIM-homeodomain transcription factor Lmx1b plays a crucial role in podocytes. J. Clin. Invest. 109, 1073-1082. Vollrath, D., Jaramillo-Babb, V. L., Clough, M. V., McIntosh, I., Scott, K. M., Lichter, P. R., and Richards, J. E. (1998). Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum. Mol. Genet. 7, 1091-1098. | |
| The transcriptional repressor protein Kid-1 |
| Several lines of evidence indicate, that transcription factors play important roles during renal development. Furthermore there are other results pointing to common aspects between renal development and acute renal failure. Based on these findings the transcription factor Kid-1 was cloned (Witzgall et al., 1993). Kid-1 belongs to a subclass of zinc finger proteins, which contain a KRAB domain at their NH2-terminus. The KRAB domain is a widely distributed and very potent transcriptional repressor domain, which may play a role in the formation of heterochromatin (Margolin et al., 1994; Witzgall et al., 1994). This is achieved by the interaction with an adapter protein which has been named KAP1, TIF-1ß or KRIP-1(Friedman et al., 1996; Kim et al., 1996; Moosmann et al., 1996). |
 |
| The KRAB domain is a widely distributed and very potent transcriptional repressor domain, which may play a role in the formation of heterochromatin. This is achieved by the interaction with an adaptor protein which has been named KAP1, TIF-1ß or KRIP-1. |
| Friedman, J. R., Fredericks, W. J., Jensen, D. E., Speicher, D. W., Huang, X.-P., Neilson, E. G., and Rauscher, F. J., III (1996). KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 10, 2067-2078. Kim, S.-S., Chen, Y.-M., O'Leary, E., Witzgall, R., Vidal, M., and Bonventre, J. V. (1996). A novel member of the RING finger family, KRIP-1, associates with the KRAB-A transcriptional repressor domain of zinc finger proteins. Proc. Natl. Acad. Sci. USA 93, 15299-15304. Margolin, J. F., Friedman, J. R., Meyer, W. K.-H., Vissing, H., Thiesen, H.-J., and Rauscher, F. J., III (1994). Kruppel-associated boxes are potent transcriptional repression domains. Proc. Natl. Acad. Sci. USA 91, 4509-4513. Moosmann, P., Georgiev, O., Le Douarin, B., Bourquin, J.-P., and Schaffner, W. (1996). Transcriptional repression by RING finger protein TIF1b that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res. 24, 4859-4867. Witzgall, R., O´Leary, E., Gessner, R., Ouellette, A. J., and Bonventre, J. V. (1993). Kid-1, a putative renal transcription factor: Regulation during ontogeny and in response to ischemia and toxic injury. Mol. Cell. Biol. 13, 1933-1942. Witzgall, R., O´Leary, E., Leaf, A., Önaldi, D., and Bonventre, J. V. (1994). The KRAB-A domain of zinc finger-proteins mediates transcriptional repression. Proc. Natl. Acad. Sci. USA 91, 4514-4518. |
| Research techniques |
|
Anita Hecht - 07.09.2021 10:44 