GW method development
Low-scaling GW method
The GW method is the state-of-the-art method implemented in scientific software to compute electronic band structures in solids and electronic level energies in molecules. Today's largest supercomputers are required, when applying GW to unit cells or molecules containing 10-100 atoms which is often required to model large molecules, liquids, interfaces or nanostructured materials. A reason for the high computational cost of GW is the unfavorable increase of computing time in GW with the fourth power of the number of atoms in the simulation, see the blue dots in the lower figure.
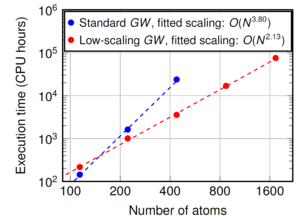 Our recent developments in the open-source software package CP2K reduced the computational cost of GW to quadratic scaling, see the red dots in the right figure. [1] We applied low-scaling GW to molecules with more than 1000 atoms. We are working on increasing the accuracy and efficiency of low-scaling GW [2] and ultimately, to properly include periodic boundary conditions.
Our recent developments in the open-source software package CP2K reduced the computational cost of GW to quadratic scaling, see the red dots in the right figure. [1] We applied low-scaling GW to molecules with more than 1000 atoms. We are working on increasing the accuracy and efficiency of low-scaling GW [2] and ultimately, to properly include periodic boundary conditions.
[1] J. Wilhelm et al., J. Phys. Chem. Lett. 9, 306 (2018).
[2] J. Wilhelm, P. Seewald, D. Golze, J. Chem. Theory Comput. 17, 1662 (2021).
Accurate and reliable GW calculations
The GW method is implemented in major quantum chemistry packages. However, different codes gave slightly different GW results for quite a long time. We have implemented GW in the Turbomole package [1]. Together with colleagues from the Fritz-Haber institute, Berlin and the University of California, Berkeley, we set up the comprehensive GW100 benchmark set [2] consisting of one hundred molecules. We were able to match results from different GW codes for best computational settings within 1 meV. In this way, we established a new standard test for getting accurate GW results for molecules.
[1] M. J. van Setten, F. Weigend, F. Evers, J. Chem. Theory Comput. 9, 232 (2013).
[2] M. J. van Setten, F. Caruso, S. Sharifzadeh, X. Ren, M. Scheffler, F. Liu, J. Lischner, L. Lin, J. R. Deslippe, S. G. Louie, C. Yang, F. Weigend, J. B. Neaton, F. Evers, P. Rinke, J. Chem. Theory Comput. 9, 5665 (2015).
Dynamical Response in the GW Approximation
In nano-sciences many scientific challenges exist that can be addressed by calculating properties of clusters and molecular systems at a quantum mechanical level. Important topics are the accurate calculation of excitation energies, the alignment of the electronic levels of subsystems, and charge transfer. Currently available approaches are either not accurate enough or not applicable to systems of nano-scale size. For solids a new Green’s function based approach hasbeen developed that can bridge this gap - the GW approach. It has proven to be a powerful tool for calculating the electronic properties of solids, but has to be adapted to quantum chemistry applications to be effective here as well.
We currently develop and implement the GW approach for calculations on clusters and molecules. This will enable the prediction of spectral functions of coupled systems, ionization potentials, and optical excitation spectra of intermediate sized, neutral and charged, systems at much higher accuracy than currently possible. By implementing this method in an efficient and widely used academic quantum chemistry package we will make it available to broad communities in quantum chemistry and computational materials sciences. We expect that the availability of this approach will have a significant impact on computational research in molecular electronics, atomic cluster physics and surface sciences.
Read more about the GW method for molecules in our review article.
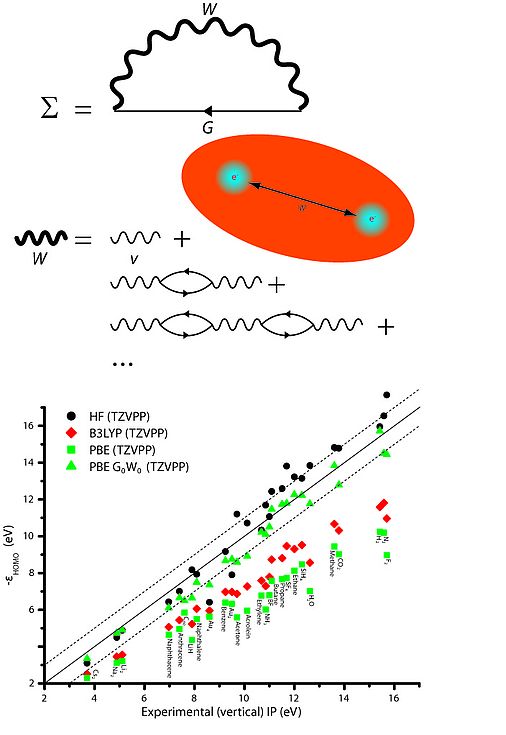
Top: The GW self-energy diagram, bottom: GW quasi-particle levels compared to experimental ionization potentials.
Webmaster Evers - 16.08.2021 13:41 
