Quantum Crystallography
Traditionally, general atomic form factors are used for structural models in crystal structure analysis, which depend only on the element and are based on simple quantum-chemically calculated electron density distributions. In contrast, quantum crystallography (external link, opens in a new window) using individual aspherical form factors is ubiquitous. These are obtained either from adapted quantum chemical calculations based on the coordinates of the present structural model or by multipole refinement. The use of these methods allows a deep insight into bonding situations and charge density distribution or even the precise determination of hydrogen positions on the level of neutron diffraction experiments. For this purpose, we mainly use the software NoSpherA2 (external link, opens in a new window), which we helped to develop.
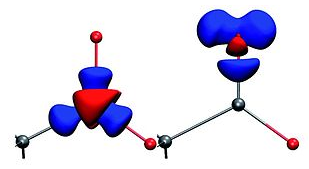
Fig. 1: The deformation density of a carboxylate group clearly shows the shift of electron density from the carbon to the bonds (left) and free electron pairs at the oxygen (right).
Cu-Kβ Radiation
Despite the significantly lower raw intensity, the use of this unusual type of radiation offers advantages over the otherwise almost exclusively used Kα radiation. For example, higher resolution ranges up to approx. 0.7 Å are accessible for copper radiation and no reflection splitting occurs at higher diffraction angles. These advantages can be seen, for example, in so-called sponge crystals (external link, opens in a new window), in which a guest molecule, partially overlaid with solvent molecules, is embedded in an organometallic framework: with Cu-Kβ radiation (external link, opens in a new window), significantly better structural models can be obtained here.
Fig. 2: The residual electron densities of a sponge crystal measured with Cu-Kα (left) and Cu-Kβ radiation (external link, opens in a new window) (right).
Anomalous Dispersion
Resonant scattering or anomalous dispersion is the inelastic interaction of the X-ray beam with the elements in a crystal structure. It is directly related to the absorption of the radiation and is basically energy and element specific. In classical crystallography, a corresponding correction is made by means of tabulated values. However, such a global adjustment is seriously wrong in some cases, which is why we developed an individual determination during the refinement (external link, opens in a new window) of the structural model. The agreement with the experimental absorption spectrum proves the usefulness of the method on the one hand and validates it at the same time.

Fig. 3: Tabulated (dotted lines) and experimental (solid lines) anomalous dispersion parameters as well as values refined by means of single crystal data (red +) and published by Sasaki (blue x) at selected energies or wavelengths.