Welcome to the Atomic-Scale Science on Insulators Group!
| |
Logo of our University formed by atomic ma-nipulation from individual copper adatoms. |
Atoms and molecules are the fundamental building blocks of matter, which is why the physical and chemical properties of these nanoscale objects govern our everyday lifes. A microscopic understanding of the world is essential for both basic research and the development of future technologies. Scanning probe methods are particularly suitable for providing insights at an atomic scale, and thanks to the continuous development of these techniques, new aspects are constantly becoming accessible.
In recent years, we contributed to such methodological development of scanning probe techniques. For example, we introduced a novel variant of scanning probe by combining principles of scanning tunneling microscopy (STM) and atomic force microscopy (AFM). As a result, we can access out-of-equilibrium charge states that are unattainable with conventional STM. Recently, we extended this method to probe spin lifetimes in individual molecules. We combined this development with electron spin resonance directly in the scanning probe microscope with atomic resolution on individual molecules. This novel approach opens up an entirely new field for investigating the spatial and temporal quantum coherence of single electrons in molecules. The strength of these combined techniques lies in their ability to relate quantum properties, such as spin lifetimes or coherences, with precise atom-scale information, transforming our microscopic perspective.
By integrating this methodological development in scanning probe microscopy with the expertise of our colleague Prof. Rupert Huber and his team, we have succeeded in combining submolecular scanning tunneling microscopy imaging with subcycle lightwave electronics. This novel development enables combined temporal and spatial resolution in the femtosecond range and on the atomic scale, opening up a completely new field of research. This approach makes it possible to investigate matter simultaneously on the relevant atomic length scales and the time scales of typical atomic and electron movements, providing an unprecedented and fascinating direct insight into our microcosm.
 | Together with Leo Gross (IBM Zurich) and Diego Peña (University Santiago de Compostela) we receive an ERC Synergy Grant for our project “Molecular Devices by Atom Manipulation” (MolDAM). |
Research Highlights
Lisanne Sellies has been awarded the 2025 Eigler Prize for her excellent talk on the work conducted in our group on ESR-AFM. Congratulations!
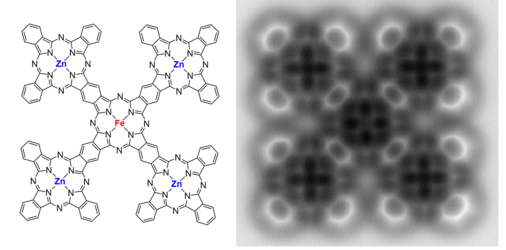
From: Angewandte Chemie Int. Edit. (2026), license CC BY-NC-ND 4.0
Combined In-Solution and On-Surface Synthesis of a Fully Fused Cross-Shaped Phthalocyanine Pentamer
Luis M. Mateo, Tzu-Chao Hung, Andreas Rank, Raffael Spachtholz, Felix Giselbrecht, Jonas Schön, Leo Gross, Jascha Repp, and Diego Peña
In this work we demonstrate a strategy for the synthesis of a fully fused, cross-shaped phthalocyanine pentamer with predefined positional control of two distinct metal species at their centers. Phthalocyanines are versatile macrocycles with attractive electronic and optical properties, yet the preparation of larger, fully fused oligomers remains challenging. By combining in-solution synthesis with on-surface reactions under ultra-high vacuum conditions, we overcome these limitations and achieve the formation of a seamlessly π-conjugated pentamer on Au(111). The resulting structure exhibits a small transport gap of 1.15 eV, reflecting the extended electronic delocalization across the fused framework.
We envision that this combined synthetic approach will enable the preparation of even larger phthalocyanine architectures through rational precursor design. In particular, two-dimensionally expanded analogues could be realized whose on-surface polymerization may give rise to molecular Lieb lattices.

Controlled single-electron transfer enables time-resolved excited-state spectroscopy of individual molecules
Lisanne Sellies, Jakob Eckrich, Leo Gross, Andrea Donarini, and Jascha Repp
In this work we found a way to access energies of charge exchange for ground and excited states of a single molecule. This allows probing many quantum transitions of different types individually, including radiative and non-radiative transitions and redox transitions, in which the charge state changes. The novel spectroscopy builds on the recently developed techniques to measure the tunneling of single electrons between a conductive tip of an atomic force microscope and a single molecule. Specifically, by slowly changing the energy of the electrons available in the tip and observing when the molecule undergoes charge-state transitions, the different excited states could be accessed, identified and their energies measured. We envision that this technique could be applied for the quantification of excitation energies that are difficult to access otherwise, for example those of triplet excitations. Similarly, it can guide the understanding of STM-induced luminescence experiments on ultrathin insulating films, for which sequential and competing transitions are extremely challenging to characterize individually.

© Brad Baxley, Part to Whole
Ultrafast atomic-scale scanning tunnelling spectroscopy of a single vacancy in a monolayer crystal
Carmen Roelcke, Lukas Z. Kastner, Maximilian Graml, Aandreas Biereder, Jan Wilhelm, Jascha Repp, Rupert Huber, and Yaroslav Gerasimenko
In this work we expanded lightwave-driven scanning tunneling microscopy combining atomic scale and sub-picosecond temporal resolution by its spectroscopy variant, now additionally providing energy resolution. Our results, thus, establish ultrafast spectroscopy on atomic length scales providing single-entity and real-space access to the ultrafast local density of states, thereby complementing techniques operating in momentum space, such as time-resolved photoemission orbital tomography. Specifically, this new combination of atomic, sub-picosecond and 10-meV-scale resolution in lightwave-driven scanning tunneling spectroscopy allowed us to observe how the energy levels of a single Se vacancy in a tungsten diselenide monolayer evolve during dynamic atomic displacements. We revealed how the excitation by a pump pulse adiabatically shifts the first defect level on picosecond timescales.
Nature Photonics 18, 595 (2024)
Titlepage of Nature Photonics issue

© Eugenio Vázquez
Single-molecule electron spin resonance by means of atomic force microscopy
Lisanne Sellies, Raffael Spachtholz, Sonja Bleher, Jakob Eckrich, Philipp Scheuerer and Jascha Repp
Routinely, molecules are imaged using atomic force microscopy (AFM), giving a visual insight into their structure. Electron spin resonance (ESR) is commonly used to characterize compounds in chemistry providing complementary information, even giving access to the isotopic composition. But ESR typically relies on measuring a countless number of molecules. In this publication, we demonstrate that we combined ESR with AFM, detecting the ESR signal via the measurement of the triplet lifetime (see below). Thereby, we could measure ESR-AFM spectra with sub-nanoelectronvolt spectral resolution in a molecule-by-molecule fashion, allowing to determine the isotopic composition of each individual molecule being measured and imaged.
Importantly, ESR relies on the manipulation of electron spin states. Due to the weakly perturbing nature of the ESR-AFM technique, we could coherently manipulate the electron spins in pentacene over tens of microseconds. The combination of atomic-scale spatial information provided by AFM and access to long coherences by means of ESR-AFM could offer insight into atomic aspects of decoherence mechanisms, potentially relevant for future quantum information processing.
see also: Contribution by J.L. Miller in Physics Today in print and online
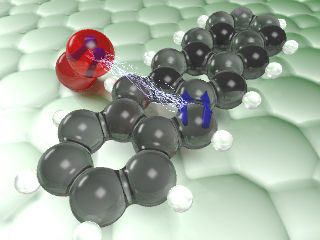
Atomically resolved single-molecule triplet quenching
J. Peng, S. Sokolov, D. Hernangómez-Pérez, F. Evers, L. Gross, J. Lupton, and J. Repp
The excited triplet in organic molecules can be quite long lived because the transition to the singlet ground state is optically forbidden. Nearby oxygen is known to quench the triplet state. We now succeeded in tracking this transfer of energy between a pentacene and an oxygen molecule directly in space. In the junction of an atomic force microscope we applied a sequence of fast electrical pulses to the pentacene molecule, driving it into the magnetic triplet state in a controlled fashion. The energy transfer from this excited triplet state to oxygen molecules nearby was then tracked in time by measuring miniscule changes in the force acting on the tip. By means of single-molecule manipulation techniques, different arrangements with oxygen molecules were created and characterized with atomic precision, allowing for the direct correlation of molecular arrangements with the lifetime of the quenched triplet.
See also: Chemistry World

Quantitative sampling of atomic-scale electromagnetic waveforms
D. Peller, C. Roelcke, L. Z. Kastner, T. Buchner, A. Neef, J. Hayes, F. Bonafé, D. Sidler, M. Ruggenthaler, A. Rubio, R. Huber & J. Repp
Visible light has a wavelength more than a thousand times larger than the size of an atom. Quite ironically, while the fundamental arrangement of the atomic world can nowadays be routinely imaged, at these atomistic length scales the behavior of light remains a mystery in many aspects. This applies in particular to the temporal behavior of light at these ultrasmall scales, where the well-known laws of classical physics lose their validity and quantum physics rules.
Together with Prof. Rupert Huber and his team as well as our theory collaborators at the MPSD in Hamburg we have developed a method to detect the dynamics of light on such a small scale with high temporal resolution. The key ingredient is a single-molecule switch acting as an atomic-scale voltage standard.

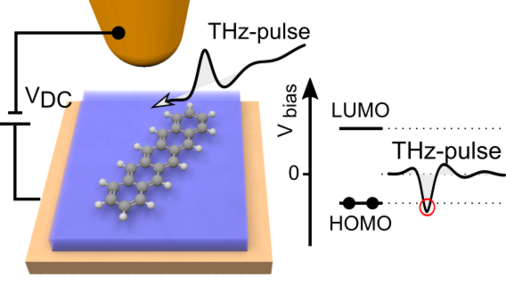
Ultrafast atomic-scale forces coherently control a single-molecule switch
D. Peller, L. Kastner, T. Buchner, C. Roelcke, F. Albrecht, N. Moll, R. Huber, and J. Repp
In 2016 in a close collaboration with Prof. Rupert Huber and his team we succeeded in combining sub-molecular STM imaging and subcycle lightwave electronics (see further below). In a low-temperature STM, an intense phase-locked half-cycle THz pulse was tightly focused onto the tunneling junction, where the electric field acts as a transient bias voltage across an STM junction.
Now, in this publication we demonstrate the combined femtosecond and angstrom access in the control of matter. The ultrafast localized electric fields in our lightwave STM enable exerting atomic-scale femtosecond forces to selected atoms. By means of these atomic forces on the intrinsic timescale of molecules, coherent atomic motion can now be excited. Utilizing coherent structural dynamics, we can modulate the quantum transitions of a single-molecule switch by up to 39%.

Implementing functionality in molecular self-assembled monolayers
Nemanja Kocić, Dominik Blank, Paula Abufager, Nicolas Lorente, Silvio Decurtins, Shi-Xia Liu and Jascha Repp
In any type of future molecular-based quantum cellular automata, the perfect placement and alignment of cells is of crucial importance, as the latter critically determines the interaction between neighboring cells. Self-assembly allow for such a precise atomic-scale alignment of zillions of molecules, however, implementing a function requires particular structures of aligned cells and not just extended monolayers thereof. We show that self-assembly can be combined with an activation of cells. In our work, selected molecules inside self-assembled islands are activated by vacancy creation from scanning-probe based manipulation, but we propose that activation can be also realized by other means as the removal of ligands, for example. This general concept shows how to combine the precise mutual atomic-scale alignment of molecules by self-assembly, on the one hand, and the implementation of specific functionality into otherwise homogeneous monolayers, on the other.
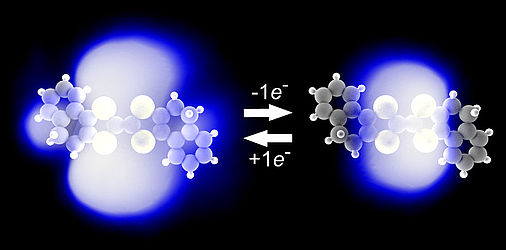
Mapping orbital changes upon electron transfer with tunnelling microscopy on insulators
Laerte L. Patera, Fabian Queck, Philipp Scheuerer and Jascha Repp
Electron transfer plays a crucial role in many chemical reactions, from photosynthesis to combustion and corrosion. However, the way in which redox reactions affect individual molecules and, in particular, their electronic structure, remains largely unclear. Unveiling these fundamental aspects requires the development of experimental tools allowing the observation of electron transfer down to the single molecule level. Here, we introduced a novel method capable of performing tunnelling experiments on non-conductive substrates to map the orbital structure of isolated molecules upon electron transfer. By driving a change in the redox state of a molecule synchronized with the oscillating tip of an AFM, previously inaccessible electronic transitions are resolved in space and energy.
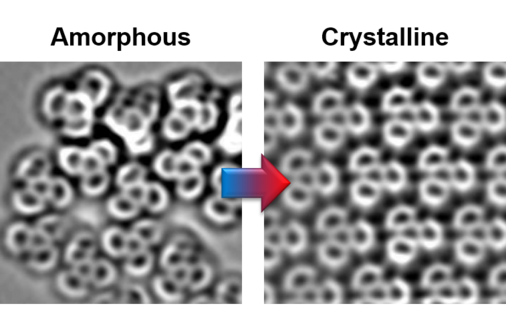
Crystallization of a Two-Dimensional Hydrogen-Bonded Molecular Assembly
L.L. Patera, X. Liu, N. Mosso, S. Decurtins, S.-X. Liu and J. Repp
We investigated the structures of a two-dimensional hydrogen-bonded film, both in the kinetically trapped amorphous state and in the thermodynamically stable crystalline phase. The sub-molecular resolution provided by nc-AFM allows resolving the changes in the bonding motifs upon transition to the crystalline state. These results reveal how the crystallization governs the length scale of the network order for non-flexible molecular species without affecting the local bonding schemes.
Angewandte Chemie International Edition 56, 10786 (2017).
Tracking the ultrafast motion of a single molecule by femtosecond orbital imaging
T. Cocker, D. Peller, P. Yu, J. Repp, and R. Huber
In a close collaboration with Prof. Rupert Huber and his team we succeeded in combining sub-molecular STM imaging and subcycle lightwave electronics. In a low-temperature STM, an intense phase-locked half-cycle THz pulse was tightly focused onto the tunneling junction. The THz field acted as a quasi-instantaneous transient bias for the tunnelling junction and the resulting time-averaged tunnelling current was measured. It was possible to record a femtosecond snapshot image of an individual molecular orbital, with a time resolution of 115 fs, determined with an autocorrelation method. By correlating two successive tunnelling events, the coherent THz vibrations of the single molecule were directly tracked in time with sub-angstrom precision.

Charge-State-Dependent Diffusion of Individual Gold Adatoms on Ionic Thin NaCl Films
J. Repp, W. Steurer, I. Scivetti, M. Persson, L. Gross, and G. Meyer
Metal atoms on insulating films can exhibit several different charge states, which can be controlled on the level of individual atoms. As an atom’s electron shell is decisive for almost all of its physical and chemical properties, a control of the charge state offers the prospect of controlling adsorbates in many other aspects.
We demonstrated that the diffusion behavior of individual gold adatoms on ultra-thin NaCl films depends strongly on its charge state and can therefore be controlled by means of STM-based manipulation.
Physical Review Letters 117, 146102 (2016).
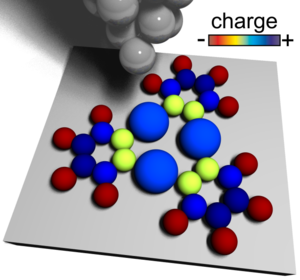
Probing Charges on the Atomic Scale by Means of Atomic Force Microscopy
Florian Albrecht, Jascha Repp, Martin Fleischmann, Manfred Scheer, Martin Ondráček, and Pavel Jelínek
Kelvin Probe Force Spectroscopy (KPFS) is an established technique based on atomic force microscopy (AFM) to determine local fluctuations of a sample’s work function. Such fluctuations can be attributed to local charges or dipoles at the sample surface. Triggered by the leap in resolution in low-temperature AFM with functionalized tips [Gross et al. Science 325, 1110 (2009)], KPFS was recently applied to study individual molecules exhibiting a dipole moment with sub-molecular resolution.
In our work we apply KPFS to successfully map out the polar nature of bonds inside individual molecules. In addition, we clarify the origin of certain artifacts in KPFS that inevitably occur at very close tip-sample distances. We introduce a new method to determine the charge distributions with sub-molecular resolution, and we benchmark this method for various systems. Thereby we demonstrate that this new method allows for resolving intramolecular charge distributions with unprecedented resolution - a regime for which conventional KPFS is shown to fail.
Physical Review Letters 115, 076101 (2015).
See also: Synopsis in Physics, Physics world, Chemistry World, COSMOS
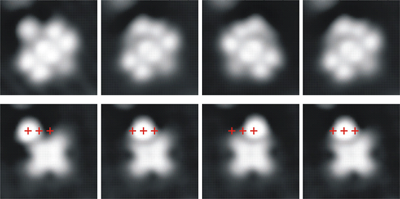
Controlling the orbital sequence in individual Cu-Phthalocyanine molecules
Christof Uhlmann, Ingmar Swart, and Jascha Repp
We investigated a molecular switch that is based on a novel concept: controlling the energetic order of molecular orbitals. This is fundamentally different from previously proposed mechanisms for molecular switching such as isomerization, charge-, or spin-state manipulation. The switching takes place in negatively charged Copper(II)phthalocyanine molecules on NaCl/Cu(100). In these, a Jahn-Teller distortion lifts the degeneracy of the lowest unoccupied molecular orbitals. By placing Au and Ag-atoms close to the molecule, the energetic order of these two levels can be switched in a bistable fashion. Hence, such a molecular switch can be actuated by subtle changes in the local vicinity of a molecule, i.e. without directly injecting charge carrier into the switch itself.

Atomic force microscopy reveals bistable configurations of dibenzo[a,h]thianthrene and their interconversion pathway
Niko Pavliček, Benoit Fleury, Mathias Neu, Judith Niedenführ, Coral Herranz-Lancho, Mario Ruben, and Jascha Repp
We have visualized a difference in the chemical structure of two molecular configurations of dibenzo[a,h]thianthrene (DBTH) molecules by means of atomic force microscopy.
Recently, Gross et al. (Science, 2009) have demonstrated the role of tip-functionalization in atomic force microscopy by resolving the chemical structure of pentacene molecules. Shortly after, this technique was used to identify the structure of an organic molecule. In this paper, we have investigated DBTH molecules adsorbed on NaCl layers in a combined scanning tunneling and atomic force microscopy (STM/AFM) study. DBTH molecules are derivates of the butterfly-shaped thianthrene molecule, in which the wings are extended by one benzene ring. By inelastic current excitations in STM mode, we can switch the wings between pointing up or down. The positions of the wings, however, could only be revealed in the AFM mode. The atomically resolved AFM images also reveal that the butterfly is flapping its wings as opposed to turning upside down.
Physical Review Letters 108, 086101 (2012).

Molecular Symmetry Governs Surface Diffusion
Tobias Sonnleitner, Ingmar Swart, Niko Pavliček, Andreas Pöllmann, and Jascha Repp
In many areas of chemistry and physics the symmetry of an object or process plays a decisive role. One prominent example is the selection rules governing optical transitions. We investigated the influence of molecular symmetry on the surface potential landscape. For this purpose, the nonthermal diffusion induced by inelastic excitations of a molecule, for which four symmetry distinct isomers exist, was studied. The observed nonthermal diffusion was found to be qualitatively different for all four symmetry distinct isomers. This demonstrates that adsorbate symmetry plays an important role in determining the surface potential landscape.
Phys. Rev. Lett. 107, 186103 (2011).
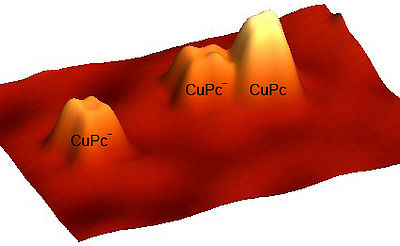
Controlling the charge state of single molecules: visualizing changes in the tunneling barrier with submolecular resolution
Ingmar Swart, Tobias Sonnleitner, and Jascha Repp
The tip of a scanning tunneling microscope was used to controllably add and remove an additional electron to and from a single molecule. It is shown that charge-state control of individual molecules adsorbed on surfaces can be obtained by choosing a substrate system with an appropriate workfunction. By subtracting images of molecules in the neutral and anionic charge state, information regarding the spatial distribution of the additional charge was obtained. These difference images show marked intramolecular contrast.
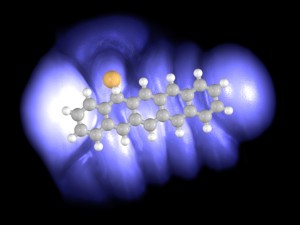
Imaging Bond Formation Between Gold and Pentacene on an Insulating Surface
Jascha Repp, Gerhard Meyer, Sami Paavilainen, Fredrik E. Olsson, Mats Persson
A covalent bond between an individual pentacene molecule and a gold atom is formed by means of single-molecule chemistry inside a scanning tunneling microscope junction. The bond formation is reversible, and different structural isomers can be produced. The single-molecule synthesis is done on ultrathin insulating films that electronically isolate the reactants and products from their environment. Direct imaging of the orbital hybridization upon bond formation provides insight into the energetic shifts and occupation of the molecular resonances.

Scanning Tunneling Microscopy Imaging of Individual Molecular Orbitals
Jascha Repp, Gerhard Meyer, Sladjana M. Stojkovic, Andre Gourdon, Christian Joachim
Ultrathin insulating NaCl films have been employed to decouple individual pentacene molecules electronically from the metallic substrate. This allows the inherent electronic structure of the free molecule to be preserved and studied by means of low-temperature scanning-tunneling microscopy. Thereby direct images of the unperturbed molecular orbitals of the individual pentacene molecules are obtained. Elastic scattering quantum chemistry calculations substantiate the experimental findings.
Phys. Rev. Lett. 94, 026803 (2005).
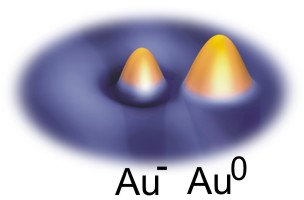
Controlling the Charge State of Individual Gold Adatoms
Jascha Repp, Gerhard Meyer, Fredrik E. Olsson, Mats Persson
The nature and control of individual metal atoms on insulators are of great importance in emerging atomic-scale technologies. Individual gold atoms on an ultrathin insulating sodium chloride film supported by a copper surface exhibit two different charge states, that are stabilized by the large ionic polarizability of the film. The charge state and associated physical and chemical properties such as diffusion can be controlled by adding or removing a single electron to or from the adatom with a scanning tunneling microscope tip.
Webredaktion - 02.03.2026 16:17 
Repp Group
Latest News

From Angew. Int. Edit. license CC BY-NC-ND, presenting a strategy that allows the on-surface synthesis of atomically precise cross-shaped Phthalocyanine pentamers.
.