News
How ATP suppresses the fibrillation of amyloid peptides: analysis of the free-energy contributions
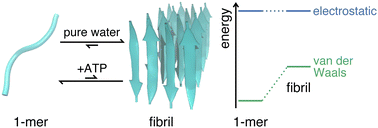
Recent experiments have revealed that adenosine triphosphate (ATP) suppresses the fibrillation of amyloid peptides – a process closely linked to neurodegenerative diseases such as Alzheimer's and Parkinson's. Apart from the adsorption of ATP onto amyloid peptides, the molecular understanding is still limited, leaving the underlying mechanism for the fibrillation suppression by ATP largely unclear, especially in regards to the molecular energetics. Here we provide an explanation at the molecular scale by quantifying the free energies using all-atom molecular dynamics simulations. We found that the changes of the free energies due to the addition of ATP lead to a significant equilibrium shift towards monomeric peptides in agreement with experiments. Despite ATP being a highly charged species, the decomposition of the free energies reveals that the van der Waals interactions with the peptide are decisive in determining the relative stabilization of the monomeric state. While the phosphate moiety exhibits strong electrostatic interactions, the compensation by the water solvent results in a minor, overall Coulomb contribution. Our quantitative analysis of the free energies identifies which intermolecular interactions are responsible for the suppression of the amyloid fibril formation by ATP and offers a promising method to analyze the roles of similarly complex cosolvents in aggregation processes.
Extended Hydrogen Bond Networks for Effective Proton-Coupled Electron Transfer (PCET) Reactions: The Unexpected Role of Thiophenol and Its Acidic Channel in Photocatalytic Hydroamidations
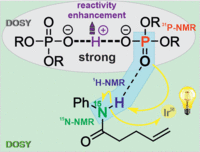 | 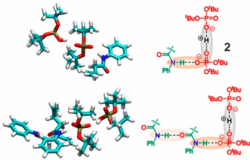 |
Preorganization and aggregation in photoredox catalysis can significantly affect reactivities or selectivities but are often neglected in synthetic and mechanistic studies, since the averaging effect of flexible ensembles can effectively hide the key activation signatures. In addition, aggregation effects are often overlooked due to highly diluted samples used in many UV studies. One prominent example is Knowles’s acceleration effect of thiophenol in proton-coupled electron transfer mediated hydroamidations, for which mainly radical properties were discussed. Here, cooperative reactivity enhancements of thiophenol/disulfide mixtures reveal the importance of H-bond networks. For the first time an in-depth NMR spectroscopic aggregation and H-bond analysis of donor and acceptor combined with MD simulations was performed revealing that thiophenol acts also as an acid. The formed phosphate-H+-phosphate dimers provide an extended H-bond network with amides allowing a productive regeneration of the photocatalyst to become effective. The radical and acidic properties of PhSH were substituted by Ph2S2 and phosphoric acid. This provides a handle for optimization of radical and ionic channels and yields accelerations up to 1 order of magnitude under synthetic conditions. Reaction profiles with different light intensities unveil photogenerated amidyl radical reservoirs lasting over minutes, substantiating the positive effect of the H-bond network prior to radical cyclization. We expect the presented concepts of effective activation via H-bond networks and the reactivity improvement via the separation of ionic and radical channels to be generally applicable in photoredox catalysis. In addition, this study shows that control of aggregates and ensembles will be a key to future photocatalysis.(https://doi.org/10.1021/JACS.0C08673)
Location and Conformation of the LKα14 Peptide in Water/Ethanol Mixtures
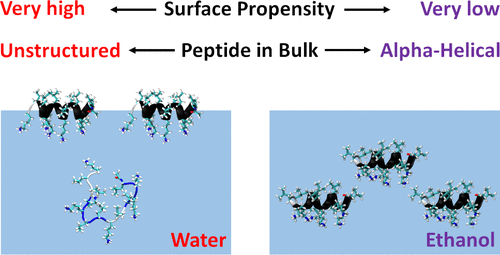
It is widely recognized that solvation is one of the major factors determining structure and functionality of proteins and long peptides, however it is a formidable challenge to address it both experimentally and computationally. For this reason, simple peptides are used to study fundamental aspects of solvation. It is well established that alcohols can change the peptide conformation and tuning of the alcohol content in solution can dramatically affect folding and, as a consequence, the function of the peptide. In this work, we focus on the leucine and lysine based LKα14 peptide designed to adopt an α-helical conformation at an apolar–polar interface. We investigate LKα14 peptide’s bulk and interfacial behavior in water/ethanol mixtures combining a suite of experimental techniques (namely, circular dichroism and nuclear magnetic resonance spectroscopy for the bulk solution, surface pressure measurements and vibrational sum frequency generation spectroscopy for the air–solution interface) with molecular dynamics simulations. We observe that ethanol highly affects both the peptide location and conformation. At low ethanol content LKα14 lacks a clear secondary structure in bulk and shows a clear preference to reside at the air–solution interface. When the ethanol content in solution increases, the peptide’s interfacial affinity is markedly reduced and the peptide approaches a stable α-helical conformation in bulk facilitated by the amphiphilic nature of the ethanol molecules. (https://doi.org/10.1021/ACS.LANGMUIR.0C03132)
Solubilization power of surfactant-free microemulsions
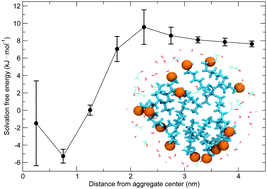
Hydrophobe solubilization concepts rely on the shielding of solutes from water in nonpolar domains comprising traditional surfactants. We show how an octanol/ethanol/water surfactant-free microemulsion solvates hydrophobic propane in small octanol/ethanol aggregates similar to traditional micelles. These aggregates have the comparable solvent quality as bulk octanol/ethanol with the same composition.(https://doi.org/10.1016/J.BPC.2019.106258)
Johnny Hioe - 16.04.2024 14:06