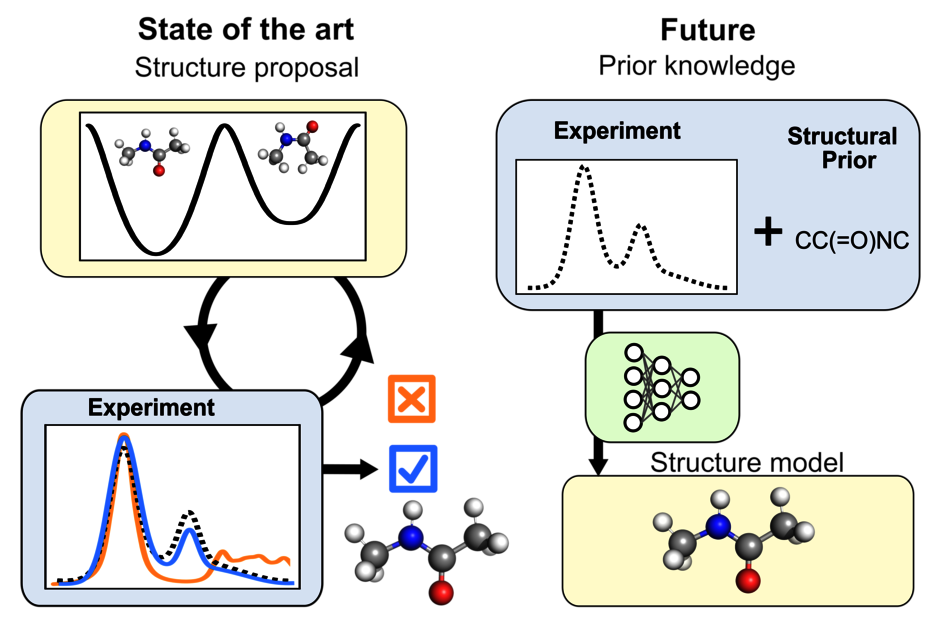
Understanding how biomolecules move and change shape is crucial to understanding their function at the atomic level. Infrared spectroscopy (IR) is a powerful method to capture these dynamic processes. Nevertheless, the translation of these spectroscopic fingerprints into high-resolution structural models remains a major challenge.
In our current work, we address this problem by predicting IR spectra directly from molecular structures obtained in biomolecular simulations. To this end, we compare two established computational approaches - normal mode analysis and Fourier transformed dipole autocorrelation function - with experimental IR data of N-methylacetamide, a model molecule for peptide binding vibrations. Force fields ranging from hybrid quantum dynamics/molecular dynamics (QM/MM) methods to machine learning and classical force fields were used.
Our results show where current computational methods already reliably reproduce experimental IR spectra and where they still fail due to structural details. These findings provide a basis for solving the "inverse problem", the direct determination of structures from experimental data.
In the future, AI-supported approaches could enable such breakthroughs and elucidate previously inaccessible biomolecule structures, such as toxic protein oligomers in neurodegenerative diseases. This would be an important step towards improved diagnostic and therapeutic approaches.
Further information can be found in our original publication:
Marvin Scherlo, Dominic Phillips, Ricarda Künne, Emiliano Ippoliti, Klaus Gerwert, Carsten Kötting, Paolo Carloni, Antonia S. J. S. Mey, Till Rudack
IR spectroscopy: from experimental spectra to high-resolution structural analysis by integrating simulations and machine learning.
The Journal of Physical Chemistry B 2025 129(45):11652-65. doi: 10.1021/acs.jpcb.5c04866 (external link, opens in a new window)