Press Releases
Admission to the Graduate Center of the bidt – early-career researcher at the University of Regensburg uses artificial intelligence to improve learning models in the health sector
September 17, 2024 Artikel auf Deutsch
“Young, smart, innovative: Today's AI talents will decisively shape our society of tomorrow. That is why we are supporting ten excellent postdocs and their AI projects with a total of around five million euros,” announced Science Minister Markus Blume in Munich today. One of the research talents receiving funding is Dr. Kata Vuk, a postdoc in the research group of Professor Dr. Merle Behr at the Faculty of Computer Science and Data Science at the University of Regensburg (UR). The mathematician's project aims to improve the application of machine learning models in the healthcare sector.
With this program, the Bavarian State Ministry of Science, Research and the Arts provides long-term support for particularly talented young AI researchers in developing their own research profile as a basis for a long-term career in science. “The fact is, artificial intelligence will significantly change our lives in many areas, and we want to shape this process according to our values,” said Minister Blume.
Starting in January 2025, researchers at the beginning of their scientific careers will be accepted into the Graduate Center at the Bavarian Research Institute for Digital Transformation (bidt), an institute of the Bavarian Academy of Sciences and Humanities. Funding is intended to last for up to four years. It includes personnel funding for the postdocs, research funding, and support for networking and further qualification in the field of digitalization. The research talents and their proposed projects were recommended for funding by a commission of experts from outside Bavaria.
Kata Vuk's project “From Data to Discovery in the Healthcare Information Age: Interpretable Machine Learning with Piecewise Constant Models” aims to improve the application of machine learning models in the healthcare sector. "I am focusing on developing models that not only enable accurate predictions but are also easy to understand. This is particularly important in the health sector, where medical decisions must be comprehensible and transparent,” explains Kata Vuk, who completed her doctorate at the Chair of Probability Theory and its Applications at the Ruhr University Bochum in 2023 after graduating in mathematics. Since June 2023, she has been strengthening Merle Behr's machine learning working group at the University of Regensburg. Her research results also flow into the Collaborative Research Center TRR 374 funded by the German Research Foundation at the University of Regensburg, which conducts research on chronic kidney disease.
Dr. Kata Vuk, University of Regensburg, is accepted into the Graduate Center at the Bavarian Research Institute for Digital Transformation (bidt). Photo © Markus Schmitt
The mathematician investigates piecewise constant models, in particular decision trees and change point models. Piecewise constant models are mathematical methods that represent complex relationships in a simple form. Decision trees and change point models help to analyze data in order to make decisions or detect changes in data patterns, for example. Kata Vuk develops approaches to adapt these models so that they can make individualized predictions while remaining comprehensible. “Decision trees, which are particularly suitable for tabular data such as patient data, offer high predictive power, but tend to lose their interpretability in complex combinations,” explains Vuk.
Change-point models, which are used for serial data such as time-dependent vital signs or genetic information, are optimized to ensure interpretability even at high dimensions: “This is crucial to identify specific changes in a patient's health status or in genetic sequences,” says Vuk. “In the long term, the project aims to use such interpretable models for personalized medicine to develop tailored treatment strategies," adds Merle Behr, who is very pleased about the success of the postdoc working at her chair.
Information/Contact
The aim of the funding line of the STMWK is to support particularly qualified young scientists in developing a research profile from the first or second year after their doctorate and thus to qualify them to remain in science. The Bavaria-wide Graduate Center is coordinated by the bidt. With its diverse research projects and dialogue activities, it offers the best conditions for the planned projects.
Dr. Kata Vuk
University of Regensburg Chair of Machine Learning
UR Research Field Digital Transformations
UR Research Field Integrated Sciences in Life, Health, and Disease
C8: The European Union is funding a project to develop a new concept of information transfer for active implanted medical devices as part of its Horizon Europe funding program
September 11, 2024 (short version) vollständiger Artikel auf Deutsch
The European Union is providing more than 3.7 million euros over a funding period of 36 months for the ERMES project as part of the highly competitive Horizon - EIC (European Innovation Council) Pathfinder Open funding program. The aim of the project is to develop a new concept for information transmission for active implantable devices (Active Implantable Medical Device, or AIMD for short). The University of Regensburg (Prof. Dr. Härteis), the Deggendorf Institute of Technology (Prof. Dr. Aung) and the FAU Erlangen-Nuremberg (Dr. M. Schäfer) are participating from Bavaria. “They were granted more than a third of the funding. A great success. Only 40 of the 1119 applications submitted this year were approved,” says Prof. Silke Härteis, who heads the Regensburg subproject. Further partners of the joint project are located in Finland and France. The Università di Catania in Italy is the lead partner. In addition to the universities, four companies are also involved.
The increase in chronic illnesses also increases the potential applications for AIMDs, which can be used to replace missing or defective body parts, administer medication, monitor bodily functions or support organs and tissues. The best-known examples of AIMDs are technical devices such as pacemakers or implantable defibrillators.
To ensure efficient communication with AIMDs, new ways of transmitting information between devices both inside and outside the body must be developed and established. In their project ERMES (INFORMATION TRANSFER BETWEEN MEDICAL DOCTORS AND IMPLANTED MEDICAL DEVICES VIA SYNTHETIC MOLECULAR COMMUNICATION), the project partners want to close this gap. Their goal is to develop novel communication and sensor concepts for AIMDs. They are building on three pillars:
1. the chemical design and synthesis of suitable messenger substances
2. the development of suitable systems to introduce the messenger substances into the body via the blood circulation system
3. the development of detection strategies for the corresponding messenger substances in the blood circulation.
Prof. Silke Härteis from the Institute for Molecular and Cellular Anatomy at the Faculty for Biology and Preclinical Studies at the University of Regensburg uses what is known as the chorion-allantoic membrane (CAM) model for this. In the CAM model, the chorion of fertilized chicken eggs – also known as the chorion-allantoic membrane – replaces traditional animal testing. It is also cost-effective and time-saving. The researcher already has experience with the model. She is involved in a sub-project of the Transregio 374 “Tubular System and Interstitium of the Kidney: (Patho-) Physiology and Crosstalk”. The collaborative project between the University of Regensburg and the Friedrich-Alexander-University Erlangen-Nuremberg aims to gain insights into the development of various kidney diseases. Using the CAM model, she is investigating the growth of a particular, severe form of hereditary renal cyst and testing promising drugs for their effectiveness.
For the project within the Horizon funding program, Prof. Härteis is using the same model, but with a completely different goal. Namely, to develop novel information systems for AIMDs in which molecules – i.e. chemical compounds – are used for communication between AIMDs within the body. This still relatively new concept of information transfer is called “molecular communication”. “Molecular communication is based on principles from nature and encodes information by using different concentrations or properties of molecules,” says Prof. Härteis.
Communication between AIMDs and external devices outside the body requires so-called gateways that make it possible to convert a signal from inside the body, such as a certain molecule concentration (MC), into a macroscopic signal such as light or electromagnetic waves. These signals are then received and processed by external devices.

Project A2: Researchers at the University Hospital Erlangen have discovered a new mechanism for cyst growth in the familial cystic kidney disease ADPKD
July 9, 2024 Artikel auf Deutsch
ADPKD is one of the most common hereditary diseases worldwide and one of the main causes of loss of kidney function and thus dependence on renal replacement therapy such as hemodialysis or kidney transplantation. The cause is the formation of numerous cysts (fluid-filled cavities) in the kidneys, which increase in size over the course of a lifetime and thus displace healthy kidney tissue. Current therapeutic approaches to slow down the growth of cysts are only effective to a limited extent and are associated with relevant side effects.
Prof. Dr. Björn Buchholz, Senior Physician at the Medical Clinic 4 (Director: Prof. Dr. Mario Schiffer) at the University Hospital Erlangen, and his team have set themselves the task of deciphering the mechanisms of cyst growth. They have now been able to identify the purinergic receptor P2Y2R, which contributes significantly to cyst growth in an ADPKD mouse model. Both the silencing of the P2Y2R gene and the pharmacological inhibition by the drug suramin significantly improved the course of the disease. "One limitation is that suramin can also inhibit other purinergic receptors and is therefore not a suitable approach for long-term therapy. However, we are pleased that we have been in contact for some time with a leading pharmaceutical company that is very committed to developing a more specific active ingredient," explains Prof. Buchholz. He adds: "The fact that knocking out P2Y2R under physiological conditions showed no significant side effects even after more than a year and that P2Y2R was also detected at high levels in the cysts of our patients makes P2Y2R a promising pharmacological target."
The work was made possible by the German Research Foundation as part of the Transregional Renal Research Network Erlangen-Regensburg TRR 374. The results were published in the renowned scientific journal JASN
Original publication:
Andre Kraus, Kathrin Skoczynski, Martin Brötsch, Nicolai Burzlaff, Jens Leipziger, Mario Schiffer, Maike Büttner-Herold, Bjoern Buchholz: P2Y2R and Cyst Growth in Polycystic Kidney Disease. Journal of the American Society of Nephrology: 10.1681/ASN.0000000000000416, June 7, 2024. DOI: 10.1681/ASN.0000000000000416.
Links:
- TRR 374(https://www.uni-regensburg.de/biologie-vorklinische-medizin/sfb1350/crc-1350/index.html)
- AG Buchholz(https://www.medizin4.uk-erlangen.de/forschung/experimentelle-forschung/mechanismen-renalen-zystenwachstums/)
Further information:
Prof. Dr. med. Björn Buchholz
Phone: 09131 85-39002
bjoern.buchholz@uk-erlangen.de.
DFG funds joint UR and FAU collaborative research with 12.3 million euros
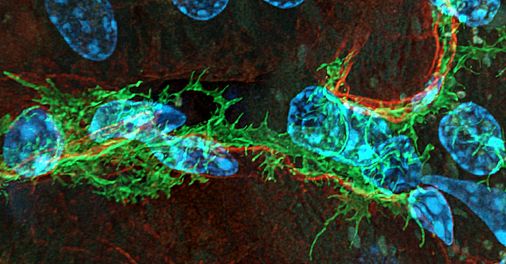
November 29, 2022
The proposal for the joint SFB/TRR 374 of the University of Regensburg (UR) with the Friedrich-Alexander-University Erlangen-Nuremberg (FAU) entitled "Tubular system and interstitium of the kidney: (Patho-) physiology and crosstalk" (formerly SFB 1350) has been approved by the German Research Foundation (DFG) for a 2nd funding period. Spokesperson is Prof. Dr. Richard Warth, Medical Cell Biology at the University of Regenburg. The total funding amount for the period from 2023 to 2026 is 12.3 million euros.
In Germany, more than 5 million patients suffer from chronic kidney disease - most of them without knowing it - and about 100,000 people require kidney replacement therapy in the form of dialysis or transplantation. For their detoxification and elimination function, the kidneys use a two-stage principle: First, a large amount of filtrate is formed from the blood plasma and then largely reabsorbed and modified in a system of tubules. Until now, research into kidney disease has focused mainly on the filtration process. The function of the tubules and the tissue surrounding them (tubulointerstitium) has hardly been studied, despite its great relevance to disease, because the interactions that take place there are exceedingly complex and methodologically difficult to address. Therefore, an interdisciplinary team of researchers has joined forces in the Renal Collaborative Research Center SFB 1350 / Transregio TRR 374 to investigate these complex processes and signaling pathways of the tubulointerstitium.
In the first funding period of the SFB 1350 from 2019-2022, the team of researchers from the University of Regensburg and the Friedrich Alexander University Erlangen-Nuremberg succeeded in gaining important insights into the development of various kidney diseases. For example, mechanisms of inflammatory processes and excessive scarring were uncovered and genetic risk factors for kidney function loss were identified.
For University President Professor Dr. Udo Hebel, the success of the Regensburg scientists is proof of the outstanding research work of the researchers involved in the SFB/TRR: "Excellent work is being done in the field of life sciences, and the extension that has now been approved is once again proof of the scientific excellence and, at the same time, the future viability of research in this field," said Prof. Hebel.
For the next funding period from 2023-2026, the research team's goal is to further deepen knowledge about the function and dysfunction of the tubulointerstitium of the kidney, to enable the development of new diagnostic and therapeutic strategies, and thus to prevent or delay the progression of kidney diseases.
From 2023, Collaborative Research Center 1350 will become Transregio 374, enabling the team led by Prof. Dr. Richard Warth, Prof. Dr. Frank Schweda, Prof. Dr. Kerstin Amann and Prof. Dr. Mario Schiffer will be able to even better combine basic research, clinical research, state-of-the-art technologies and data science, and further synergies can be created between the complementary eastern Bavarian kidney locations of Regensburg and Erlangen.
"The DFG's positive funding decision confirms our successful work and puts us, as Transregio 374, in an ideal position to shape modern kidney research together with national and international partners, train the kidney researchers and kidney doctors of the future, and develop tailored diagnostics and therapies for kidney disease patients," said Prof. Warth. "This success," Warth continued, "is also the moment to say thank you. The team of spokespersons would like to thank the researchers for their outstanding work and team spirit, but also especially for the great support from the two university administrations, the ministry and the colleagues of the research funding structures in Regenburg and Erlangen. In an ever tighter competition, in which a blink of an eye can make the difference, everyone involved has to pull together. This is the only way we were able to achieve this success."
Projects A2 and A3: Research teams from Regensburg and Erlangen find a promising approach to the treatment of the familial cystic kidney disease ADPKD
August 28, 2020 Artikel auf Deutsch
"Cystic kidneys" are one of the most common inherited diseases worldwide, requiring dialysis in advanced stages. This is caused by the appearance of cysts (fluid-filled cavities) in both kidneys, which continuously increase in size and thus displace healthy tissue. The therapeutic options available to date are only very limited in their effectiveness and are accompanied by side effects. Researchers at the University of Regensburg and the University Hospital Erlangen have now shown in a comprehensive study that the chloride channel TMEM16A (anoctamine 1) contributes significantly to cyst growth and that pharmacological inhibition of TMEM16A significantly reduces cyst growth. This is achieved in part by two drugs already approved for other purposes in human medicine. The results have now been published in the journal Nature Communications.
About one in a thousand people suffer from the autosomal dominant polycystic kidney disease ADPKD, which in turn leads to irreversible loss of kidney function in about 50% of cases from the middle of the fifth decade of life. For patients, this means lifelong hemodialysis or kidney transplantation and a shortened life expectancy. In addition, those affected suffer from high blood pressure, pain and infections as a result of their kidney disease. It has been known for some time that continuous cyst growth is a major contributor to all these problems. The therapeutic options available to date have only very limited efficacy and are accompanied by relevant side effects.
An essential mechanism for cyst growth is the transport of fluid into the interior of the cysts. The research teams led by Professor Dr. Karl Kunzelmann and Professor Dr. Rainer Schreiber from the Institute of Physiology at the University of Regensburg and PD Dr. Björn Buchholz at the University Hospital Erlangen now report a potential breakthrough: Their research groups show that genetic inactivation of the chloride channel TMEM16A in an ADPKD mouse model led to a significant reduction in cyst growth. In a next step, they found that the highly specific pharmacological agent Ani9 achieved comparable inhibition of cyst growth. In their goal toward treating patients with renal cysts, they identified two promising compounds that suppressed cyst growth in mice and in cell experiments. These are benzbromarone, which has long been used to lower uric acid, and niclosamide, also an approved drug.
In their joint publication, the authors reveal the precise mechanism of action leading to cyst formation and its pharmacological inhibition: "Further studies are now needed to show good efficacy and tolerability in patients as well." In this context, the researchers refer to the Collaborative Research Center 1350 "Tubule System and Interstitium of the Kidney: (Patho-) Physiology and Crosstalk," in which they are involved with their research groups: "Collaborative Research Center 1350 offers us optimal conditions for conducting the experiments and for our close collaboration," Karl Kunzelmann and Björn Buchholz agree.
Original publication:
Ines Cabrita, Andre Kraus, Julia Katharina Scholz, Kathrin Skoczynski, Rainer Schreiber, Karl Kunzelmann, Björn Buchholz: Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. In: Nature Communications.
DOI http://dx.doi.org/10.1038/s41467-020-18104-5

Project A2: The role of a gene in the kidney allows a simple diagnostic method to be developed
July 29, 2020 Artikel auf Deutsch
Cystic fibrosis is primarily a severe lung disease, but it also affects other organs, such as the pancreas and intestines. Cystic fibrosis is caused by mutations in the so-called CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene. This gene codes for a chloride channel that is typically found in organs that need to transport a lot of salt. The kidney is also such an organ. It is therefore all the more surprising that the kidney does not appear to show any functional impairment in the disease cystic fibrosis. The research teams led by Prof. Jens Leipziger in Aarhus/Denmark and from the Regensburg laboratory of the research duo Prof. Dr. Karl Kunzelmann and Prof. Dr. Rainer Schreiber have now made significant progress in understanding the role of CFTR in the kidney as part of Collaborative Research Center 1350.
CFTR is mainly found in specialized cells of renal tubules that help to adjust the pH of the blood. The paper by the Aarhus and Regensburg scientists, which has now been published in the Journal of the American Society of Nephrology, describes the precise cellular mechanism of pH regulation and the central role played by CFTR in this process. CFTR is activated by the digestive hormone secretin, which intervenes in a regulatory manner. One would now expect to find conspicuous changes in blood pH on a regular basis in cystic fibrosis patients. However, this is not the case, since other parts of the kidney and, above all, respiration are in the foreground in blood pH regulation in healthy people. However, an elevated pH, a so-called alkalosis, has been repeatedly reported in cystic fibrosis patients.
Currently, the diagnosis of cystic fibrosis disease is not straightforward because there are well over 2,000 different mutations. Current diagnostic procedures are either inaccurate, laborious, or burdensome and usually do not directly test CFTR function or residual function. The fact that cystic fibrosis patients cannot adequately regulate blood pH in the presence of alkalosis gave the researchers the idea of using this to diagnose cystic fibrosis: "The sick children or even older patients would simply have to drink a beverage that raises the blood pH for a short time. An increased pH value and the excretion of so-called bases could then be detected in the subsequently excreted urine," explains Prof. Leipziger. "If cystic fibrosis is present, this excretion would be absent," adds Prof. Kunzelmann. Precisely these findings have now been demonstrated in animal models of cystic fibrosis and in cystic fibrosis patients. Furthermore, the researchers were able to show that such a simple drinking test can be reliably used as a success control for therapy with recently developed CFTR repair drugs, so-called CFTR modulators. Such a simple test could also help save costs of the very expensive therapy. "As a next step, we are planning clinical trials to assess the everyday suitability of this simple and cost-effective cystic fibrosis test," the team is already looking ahead.
Original publication:
Peder Berg, Samuel L. Svendsen, Mads V. Sorensen, Casper K. Larsen, Jesper Frank Andersen, Soren Jensen-Fangel, Majbritt Jeppesen, Rainer Schreiber, Ines Cabrita, Karl Kunzelmann and Jens Leipziger: Impaired renal HCO3- excretion in Cystic Fibrosis, Journal of the American Society of Nephrology
DOI: https://doi.org/10.1681/ASN.2020010053
RW - 20.09.2024 12:27 
MISSION
"Interdisciplinary kidney research to advance understanding of disease mechanisms and develop new therapeutic concepts"

Contact:
Dr. Michaela Kritzenberger
Email
Tel.: ++49 (0)941/943-2885