Cell Cloud Analysis
The presence or absence of specialized immune cells can cause disease or affect treatment response. If the characteristics of these cells are unknown, artificial intelligence can help defining and characterizing these critical cells. We develop an automated analysis pipeline based on convolutional networks already in use in the field of point cloud classification to (a) identify these cells from flow cytometry data and (b) to predict treatment response.This project is carried out in close collaboration with INsTRuCT.
Metabodefense: The host metabolism as an antimicrobial effector
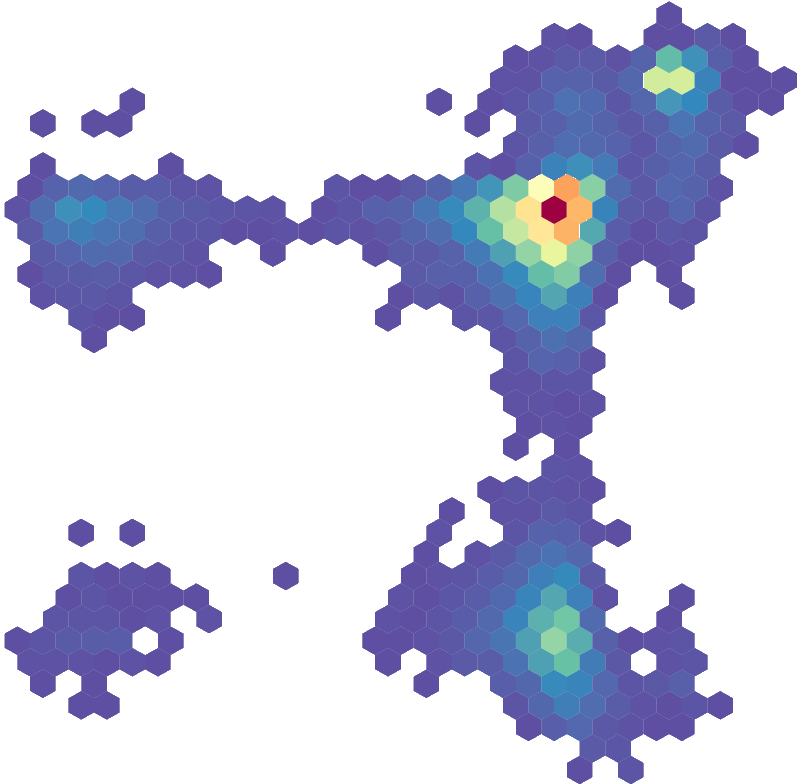
Products of the body’s own metabolism not only have a regulatory effect on the immune system but can also influence the growth or persistence of bacteria. Metabodefense aims at finding antimicrobial structures within the host metabolism that enhance pathogen control. To this end, we apply machine learning techniques to predict pathogen control and causal discovery algorithms to identify potential molecular targets. Furthermore, we work on preprocessing methods for high-throughput metabolic data sets based on Mixed Graphical Models which can be used to detect anomalous data points in high dimensional data. This project is part of bayresq.net (externer Link, öffnet neues Fenster)
AI assisted lymphoma pathology
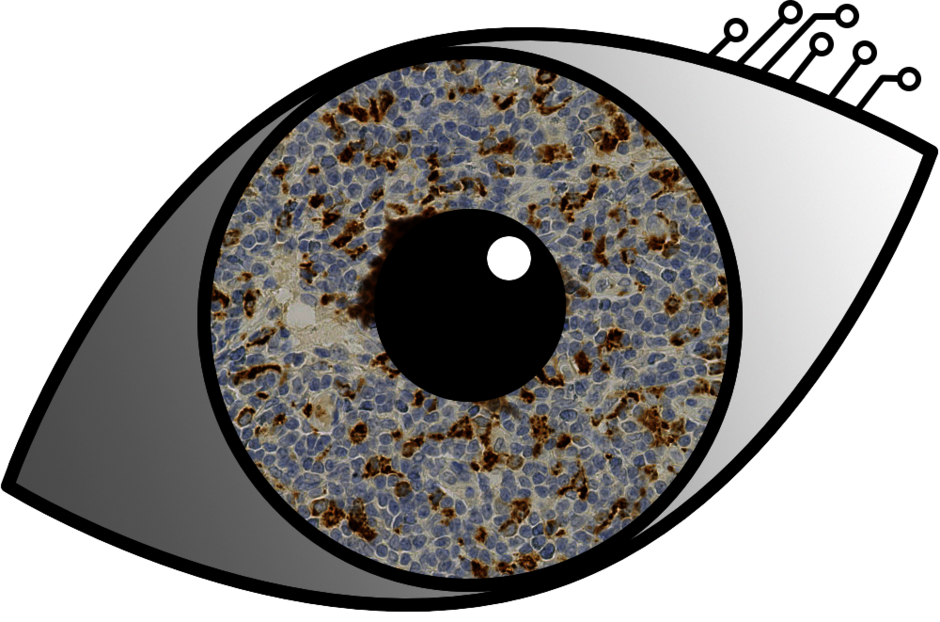
Oncologic precision medicine needs very exact sub-typing of tumors to enable the best possible therapy response. While pathologist are very good at this, diagnosing these tumors is a very laborious and analog process with limits in how many patients can be diagnosed, how much area of the slides can be looked at and how many subtypes are distinguishable by the human eye. But augmenting this process through digitalisation is possible, slides can be scanned digitally as images with about 10 gigapixels resolution. We are developing a solution to automatically diagnose different subtypes of lymphoma or benign lymphadenitis based on these high resolution scans of lymph node slides. We use deep convolutional neural networks to train a pathology assistance tool based on a data set of 628 scanned and labeled slides from 157 patients with 4 different immunohistochemistry stains each.
Zero-Sum Regression
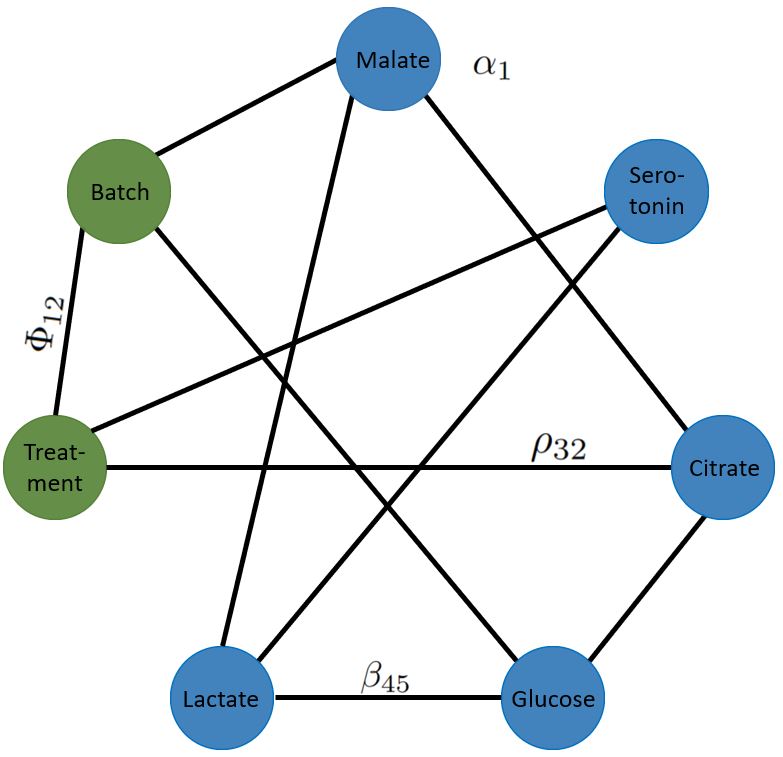
Molecular measurements are relative to a reference point, like a fixed aliquot of RNA extracted from a tissue, a defined number of blood cells, or a defined volume of biofluid. Reference point discrepancies across data sets compromise the performance of regression models like the LASSO. We have developed zero-sum regression for a reference point insensitive analysis that enhance cross-platform analysis. An R-package implementation can be downloaded here (externer Link, öffnet neues Fenster)
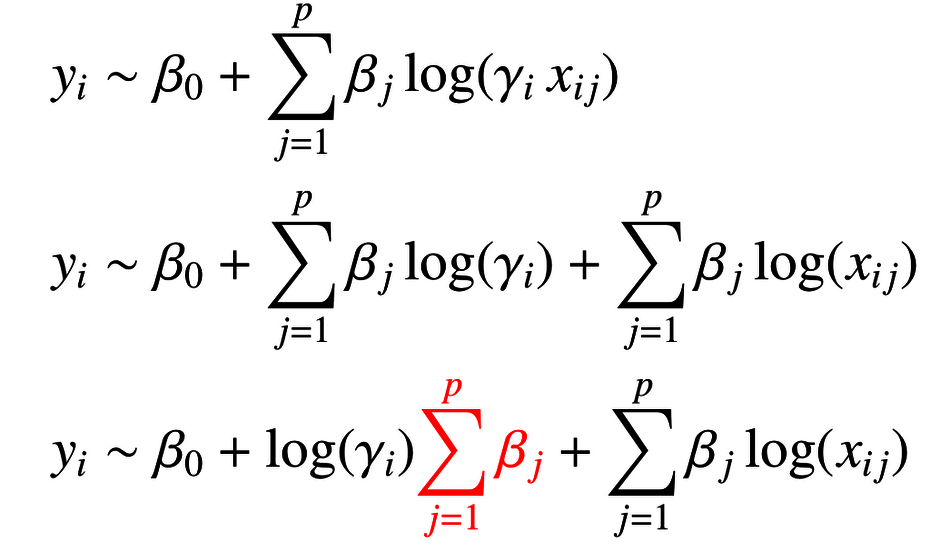
TissueResolver - a new algorithm to predict cell type specific gene expression in tissues
From the many available single cell profiles publicly available, we find and combine a small number of "prototypic" cells that are best suited to describe RNASeq data of bulk tissue, allowing us to attribute gene expression to certain sub-populations of cells and address the question "what are the individual cells in the tissue doing?". This approach preserves the biological complexity of cells by avoiding model assumptions but turns the problem into a combinatorial machine learning problem that is NP-hard and requires smart and efficient algorithms."
